药物监管行业的起源(药物开发系列 3/6)
在本系列的前几篇文章中,我们了解了参与药物开发的人员以及将药物推向市场所需完成的工作。人们不禁要问——为什么药物开发需要如此严格的程序、大量的时间和资金?了解药物开发的历史或许能提供背景和前瞻性,帮助我们看清其当前和未来的方向。当你审视这段历史时,你会注意到变革往往被归因于对悲剧的反应,尽管在许多情况下,更深入的考察会发现改革其实早已在进行中。
我们还将看到,随着国际协调法律的努力取得成功,不同国家的监管历史开始重叠,特别是在 20 世纪下半叶。请注意,以下内容主要从美国视角概述——我们鼓励读者探索 Dan Carpenter 对 FDA 的历史分析以获得更详细的理解,以及其他作者关于各管辖区域的著作。
美国:追溯药物监管的起源
我们当时还不知道,但在 1900 年代购买药物就像掷骰子一样。当时,许多”专利药”声称能治愈从头痛到心痛的一切疾病。例如,D. Jayne 博士的补虫剂在 1896 年被宣传为治疗喉咙和呼吸系统疾病的万能药和驱虫药。

D. Jayne 博士补虫剂的广告
19 世纪专利药的另一个标志性代表是”蛇油”,被吹捧具有从治愈头痛到缓解慢性疼痛的所谓治疗功效。蛇油的传说象征着典型的专利药,体现了在严格的药物监管和测试出现之前,那个时代普遍存在的未经验证的宣称和安慰剂疗法。

Clark Stanley 在 1800 年代著名的”蛇油”传单和瓶子照片
这就是药物监管出现之前的景象。想象你是这段叙事中的一位科学家,渴望测试和销售你的新药。也许这种缺乏监管的环境会让你感到自由,但随着时间的推移,你开始看到标准化和安全协议的关键需求是如何变得显而易见的。随着时间的推移,不断演变的监管框架将越来越多地影响你的工作。
自 1880 年代以来,人们一直在努力规范食品和药品的正确标签并防止两类产品中的污染。未受监管的食品和药品造成的危害非常普遍,例如受污染的白喉抗毒素事件导致多名儿童死亡。《生物制品控制法案》(1902年)首次在美国将检查和纯度测试制度化,标志着向确保药物安全性和有效性的正式监管迈进。此后不久,美国医学会(AMA)率先开展自律性药物批准计划,要求公司在 AMA 期刊上做广告之前提供其药物疗效证据。这些为后续的监管措施树立了先例,如美国食品药品监督管理局(FDA)的《纯食品和药物法案》(1906年),使销售受污染的食品或肉类成为违法行为。该法律要求如实标签——至少目标是没有人能再在标签上”画大饼”。
美国:反应性、主动性和监管措施
许多法规源于公共卫生灾难后引发的公众愤怒。
-
《联邦食品、药品和化妆品法案》(1938年):要求在批准前证明安全性,是在一起配方不当的抗生素导致超过一百人死亡的惨痛悲剧后引入的。
-
良好生产规范(GMP,1940年代初):在一批磺胺类药物被镇静剂苯巴比妥污染导致出问题(造成数百人死亡和受伤)之后制定。
-
1947年《纽伦堡法典》:作为临床试验的指导方针(确立了自愿同意等伦理研究原则),是在纳粹德国进行的非人道实验之后出台的,后来被 1964年《赫尔辛基宣言》所发展。
-
FDA 禁止 GAS 疫苗(1979年),这是一项可以说是过度反应的禁令,禁止在疫苗中使用 A 族链球菌(GAS)及其衍生物,原因是 1969 年的一项研究中 21 名健康受试者中有两到三人在接种疫苗后出现风湿热。尽管 M 蛋白疫苗——针对 GAS 等细菌成分——有着成功的长期历史,但 FDA 的禁令有效地抑制了该领域近三十年的研究。鉴于与 GAS 相关的高度未满足医疗需求,这一决定被认为特别重大。直到 2006 年禁令才被解除,允许恢复重要的研究和前瞻性突破。
-
**更快获取救命药物(1990年代):**尽管受到谨慎监测,但在 AIDS 活动人士批评 FDA 在 1990 年代阻碍获取潜在救命药物和药物审批缓慢之后,获取救命药物的渠道得到了改善。
-
FDA 性别指南(1993年)**:**要求在两性中测试药物,推翻了 1977 年指南中将育龄女性排除在早期临床研究之外的规定(该规定是对下文描述的沙利度胺灾难的回应)。
然而,重要的是要注意,虽然人们很容易认为药物监管纯粹是公众愤怒的产物——一种对高调灾难的立法应激反应——但这只是部分事实。许多改革在悲剧”加大火力”之前就已经在酝酿之中。此外,值得注意的是政策演变从反应性向主动性监管的转变。如果说立法变化过去更多是由危机驱动的,那么今天显然有一种努力在预见风险和实施预防措施。自愿性指南也发挥了作用,有时源自行业最佳实践,后来成为监管期望。
-
早在 1933 年,为了展示 1906年《纯食品和药物法案》的不足并说明需要新法律,FDA 推出了**“美国恐怖室”**展览,展出了有问题的产品,如不安全的医疗器械(如有害的子宫托和致命的减肥药)和有毒化妆品。第一夫人埃莉诺·罗斯福参观了该展览后,利用它推动了更强有力的消费者保护。这是 FDA 主动推动立法变革的一个明确实例。
-
FDA”批次认证”**(1940年代初):**对某些关键药物实行强制认证,从 1941 年的胰岛素开始,然后是 1945 年的青霉素。公司被要求在产品投放市场前从每个批次中提交样品供 FDA 检测。该政策后来扩展到所有抗生素,但在 1983 年停止执行——这是法规变得不那么严格的罕见例子。
-
《贝尔蒙特报告》(1979年):为人类受试者研究制定了伦理标准,强调尊重人格、行善和公正。如果你是科学家,你现在有责任确保药物发现和临床测试的每一步都遵守这些伦理准则——这是该领域不断演进的诚信的证明。它深刻影响了美国的临床研究法规,指导着医学试验中的伦理考量。大约在同一时期,其他国家的生物伦理委员会也取得了进展,发布了类似的指南,加强了全球对伦理标准的承诺。
沙利度胺灾难(1950年代末 - 1960年代初)是药物开发历史中的重要篇章,突出了反应性和主动性之间历史张力的重要性。沙利度胺——一种最初被批准用于治疗孕妇晨吐、在 1950 年代广泛使用的药物——导致了严重的先天性缺陷,这些在 1960 年代变得明显,引发了公众对更强药物法律的呼声。美国参议员 Kefauver 已在倡导药物改革,他抓住这一危机来重振在国会中停滞不前的法案。1962 年 7 月,Kefauver 公开了该药物的有害影响,为他的法案注入了新的活力——他觉得该法案已经失去了动力。
除了引发大量反应外,这场悲剧催化了美国和全球范围内药物监管的广泛改革。在美国,这场灾难导致了一项要求任何计划进行药物临床研究的申办方向 FDA 提供详细研究大纲并证明其疗效(而不仅仅是安全性)的法规。在国际上,该事件推动了欧盟建立新药评估的集中授权程序。例如,英国通过了 1968年《药品法案》,建立了全面的药物分类体系。总体而言,沙利度胺灾难是对该行业的一记警钟,催化了不仅是反应性的,而且是全面且强有力的改革。
药物开发的全球协调
国际协调会议(ICH)从 1990 年代开始致力于标准化药物开发过程(类似于 USB-C 试图标准化连接器的方式)。ICH 倡议代表着朝着更精简和统一的全球药物开发范式迈出的集体步伐,开创了跨国药物制造商协作努力的新时代。通过汇集来自欧洲、日本和美国的监管机构,ICH 确保在一个地区测试和批准的药物可以更容易地在其他地区获得认可,从而加速全球获得重要药物的机会。
引用他们的网站:“ICH 的使命是在全球范围内实现更大的协调,以确保安全、有效和高质量的药物以最节约资源的方式开发和注册。协调是通过 ICH 指南的制定过程实现的,该过程由监管和行业专家携手合作,通过科学共识达成。”
ICH 已经开发了广泛的新协调、澄清、修订和维护流程,并拥有约 150 份指南文件,涵盖安全性、有效性、质量和其他方面。在 ICH 流程的第 5 步,协调后的 ICH 指南由 ICH 监管成员和观察员在各自的国家/地区内实施。各监管成员和观察员国家/地区对 ICH 指南的实施和遵守情况通过独立第三方调查进行监测(参见 2021 年项目报告)。ICH 共有 21 个成员组织,每个组织有两名代表。许多监管当局将 ICH 指南纳入国内法律,使合规成为法律要求。
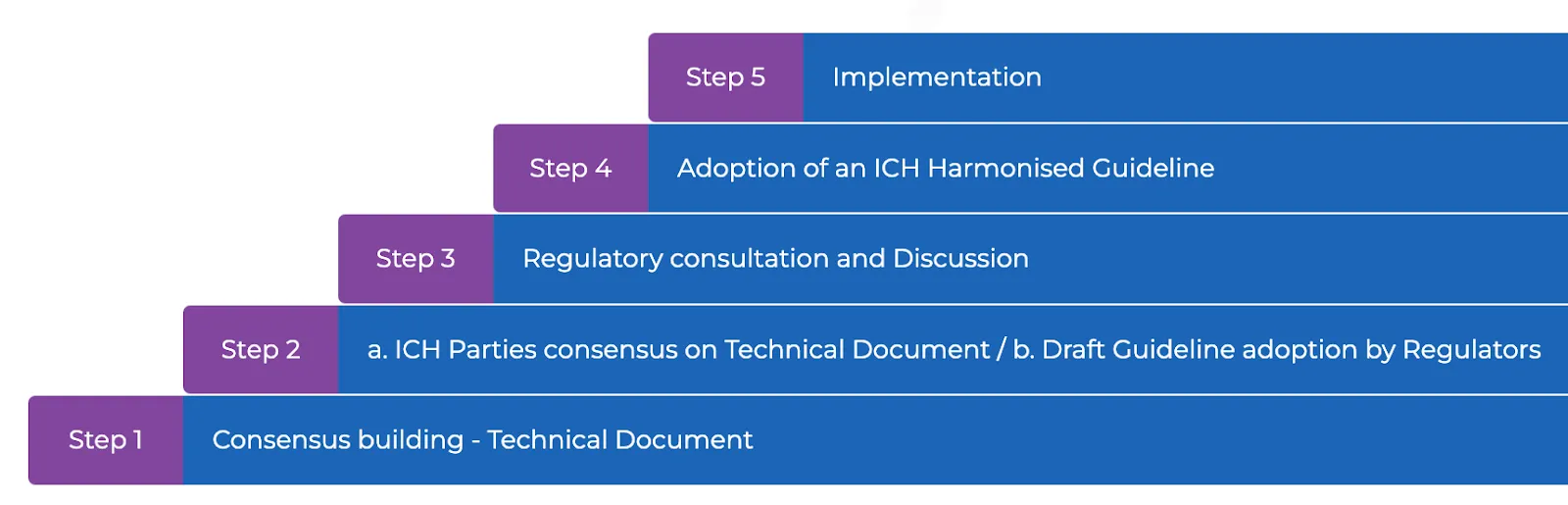
ICH 流程。
ICH 流程运作的一个例子:进入 2023 年,ICH 的良好临床规范(GCP)指南正在进行持续修订。它们于 1996 年启动,最后一次更新于 2016 年,最新修订版目前可作为草案获取。这些指南持续演变,关注新的试验类型和数据来源,反映了监管机构和行业保持前瞻性的承诺。
全球监管里程碑:从北京到俄罗斯的巡礼
虽然许多西方国家的历史与美国大致相似,但本节对跨国界和海外情况进行了非常简短的一瞥;药物监管的叙事在不同国家也展开着独特的篇章。每个国家都有其历史事件和改革,塑造着各自的监管格局。本节旨在提供简要概述,并从以美国为中心的药物开发视角中转移出来——我们链接了深入了解特定国家的资料来源,并不声称对这些地区有深入的专业知识。让我们探索一些国家的几个关键里程碑。
巴西
-
国家卫生监测局或 ANVISA(1999年)**:**成立旨在通过卫生控制保护国民健康。
-
第 13,411 号法律(2016年):修改了药品监管环境,降低了临床研究中的官僚壁垒,改善了药品注册和上市后监测流程。
-
延伸阅读:巴西的药品注册
中国
-
**中国医药公司(1950年):**被指派负责全国药品批发业务,当时医院/诊所正从私营部门转为政府直接控制。
-
GCP 启动,由国家药品监督管理局(SDA)实施(2001年)
-
CFDA 改革(2015年)**:**由中国食品药品监督管理总局宣布,旨在加速药品审批流程,让中国患者更快获得新药。
-
**ICH 成员资格(2018年):**中国成为 ICH 的正式成员,展示了其将药品法规与全球规范协调的承诺(以及 ICH 的影响力)。
-
延伸阅读:中国药物监管史
印度
- 监管前时代
传统印度医学体系的实践(20世纪前)使用阿育吠陀、悉达和尤纳尼等天然成分进行治疗。
-
**进口时代(20世纪)**殖民时期大多数药物从欧洲进口。
-
印度早期制药业始于 20 世纪初少数本土公司生产基本药物,限制了当地制药业的发展。
-
《药品和化妆品法案》(1940年)**:**对印度药品和化妆品的进口、生产、分销和销售建立了监管控制,使销售不合格药品成为严重违法行为。
1945 年,政府制定了《药品和化妆品规则》,对药品按给定的”附表”进行分类,并为药品的储存、销售、展示和处方提供指南。
-
1988 年,该法案经过改革,整合了 WHO 的 GMP 原则。
-
2005 年,该法案的”附表 Y”(提供临床试验指南的附表)更新,纳入了在印度进行临床试验的更精确标准和说明。
-
**《专利法案》(1970年):**通过只承认工艺专利而非产品专利,允许了印度仿制药行业的发展。
-
现代化和全球扩展(2000年代至今):
成为”合同研究和生产服务”(CRAMS)领域的重要参与者
-
成立监管机构如中央药品标准控制组织(CDSCO)和国家药品定价局(NPPA),以提高药品质量和安全标准并确保可负担性
-
由于引入世界贸易组织与贸易有关的知识产权协定(TRIPS),印度专利法发生了变化,影响了仿制药生产的方式。
-
《新药和临床试验规则》(2019年)**:**改革了监管框架,降低了临床研究的复杂性,改善了药品注册和上市后监测流程。
俄罗斯
-
苏联时期的监管(1917年革命后 - 1930年):1917年革命后,政府建立了国家垄断,控制药品生产、分销和质量保证的所有方面,包括 1923 年成立药典委员会(控制药品质量),以及到 1930 年人民卫生委员会全面管理药品活动。
-
**近期监管(2010-2011年):**颁布联邦法律,旨在改善医疗保健和药品供应,以及 2010 年通过”关于药品流通”法律,以确保药品的质量和安全,反映了不断发展的医疗保健格局。
津巴布韦
-
《药品及相关物质控制法案》(1969年):一部开始对该国药品进行系统性控制和管理的法案。
-
津巴布韦药品控制局或 MCAZ 的成立(1997年):成立旨在监督和管理该国的药品。
-
《顺势疗法药品法规》(2015年)**:**一项法案,代表正式撤销津巴布韦此前关于顺势疗法药品的一套法规。
-
良好分销规范(GDP)指南的采用(2018年):近期,MCAZ 采用了 GDP 指南,以确保医药产品在分销过程各环节的质量和安全。
没有独立药物监管体系的国家怎么办?
尽管有这些策略,研究表明发展中国家在采用药品质量和监管基础设施的全球标准方面仍面临阻力。没有自己独立药物监管体系的国家(通常是没有自己明确监管当局的较小国家)采用以下策略: