
Der Bauplan der Arzneimittelentwicklung (2/6 Artikelserie zur Arzneimittelentwicklung)
Anmerkung der Autoren: Diese Serie wurde in Zusammenarbeit mit James Smith und Kirsten Angeles verfasst.
Überblick
In unserem letzten Beitrag haben wir uns die verschiedenen Akteure in der Arzneimittelentwicklung und ihre Rollen bei der Markteinführung eines Medikaments angesehen. Um die akribische Natur des Weges dieser Akteure zu würdigen, schauen wir uns den eigentlichen Prozess an, an dem sie teilnehmen, von der Idee bis zum Medikament.
Es ist allgemein bekannt, dass die Arzneimittelentwicklung über mehrere Phasen verläuft und sorgfältigen Protokollen und Dokumentationen vor der Marktzulassung unterliegt. Obwohl Unternehmen ihre F&E-Budgets nicht offenlegen, schätzen Analysen die Kosten für die Markteinführung eines neuen Medikaments auf weniger als 1 Milliarde bis über 2 Milliarden Dollar. Darin enthalten sind die Kosten des Scheiterns; etwa ein Drittel dieses Budgets wird für Kandidaten ausgegeben, die es nie auf den Markt schaffen. Die Entwicklung eines neuen Medikaments ist nichts für schmale Budgets.
Im Folgenden schlüsseln wir nicht nur die Hauptphasen auf — Herstellung, Entdeckung, präklinische Tests, klinische Tests und Marktzulassung — sondern möchten auch einen Blick hinter die Kulissen mit Beispielen geben. Wir gehen auch auf einige der verschiedenen Good-Practice-(GxP-)Leitlinien ein — d. h. Good Manufacturing Practices (GMP), Good Laboratory Practices (GLP) und Good Clinical Practices (GCP).
| Phase | Kernaspekte | Durchschnittliche Dauer / Daten | Wichtige regulatorische Leitlinien | Kosten und Investitionen |
|---|---|---|---|---|
| Herstellung | GMP-Konformität, Qualitätssicherung, Dokumentation | N/A | GMP (Good Manufacturing Practices) | 15 % des F&E-Budgets; ein- bis zweistellige Millionenbeträge |
| Entdeckung | Wirkstoff-Screening, In-vitro-, In-silico-Tests | Jahre | Biosicherheitsstandards | Ein Drittel der Gesamtausgaben für gescheiterte Kandidaten |
| Präklinische Forschung | Pharmakokinetik, Toxikologie, Pharmakodynamik, Wirksamkeit | ~31 Monate im Durchschnitt | GLP (Good Laboratory Practices) | ~75.000 $ für eine einzelne Studie mit 100 Mäusen, variiert stark |
| Klinische Entwicklung | Phase-I- bis -IV-Studien, Studienbetrieb | 5,9 bis 13,1 Jahre (variiert nach Arzneimitteltyp) | GCP (Good Clinical Practices), IND-Einreichung | ~5-30 Mio. $ pro Studie im Durchschnitt — variiert stark |
| Marktzulassung | NDA/BLA-Einreichung, FDA-Prüfung | 6-10 Monate für Erstprüfung, oft mit Nacheinreichungen; ~400 IND-Anträge/Jahr; 38 Neuzulassungen/Jahr | NDA (New Drug Application), BLA (Biologics License Application) | Ca. 2-3 Mio. $ als Anmeldegebühr |
| Post-Marketing-Überwachung | Berichte zur Post-Marketing-Überwachung, Wirksamkeitsstudien | Berichte anfangs alle 6 Monate, verlängernd auf 3+ Jahre | Berichte zur Post-Marketing-Überwachung | N/A |
Herstellung
Herstellung wird von der ICH definiert als “alle Vorgänge des Materialempfangs, der Produktion, Verpackung, Umverpackung, Kennzeichnung, Umkennzeichnung, Qualitätskontrolle, Freigabe, Lagerung und des Vertriebs von [Arzneimitteln] und der damit verbundenen Kontrollen.”
Der entsprechende globale Standard, der vorschreibt, wie Medikamente hergestellt werden sollen, und sicherstellt, dass Qualität in jeden Schritt des Herstellungsprozesses “eingebaut” wird — weit über die Prüfung des Endprodukts hinaus — heißt GMP. In den USA heißt er Current Good Manufacturing Practices (cGMP), wobei betont wird, dass Hersteller aktuelle Technologien und Ansätze verwenden müssen, um konform zu bleiben.
In der Praxis bedeutet dies, dass es Anforderungen für jeden denkbaren Teil der Herstellung gibt — schauen Sie sich diesen GMP-Leitfaden an, um einen Eindruck zu bekommen — Sie werden sehen, dass selbst die Personalhygiene ein paar Zeilen hat. Die Einhaltung der geforderten Prozesse umfasst schriftliche Protokolle (z. B. in Form von SOPs, Herstellungsvorschriften), Mitarbeiterschulungen, interne Audits, Gerätetests, Verwendung hochwertiger Materialien (z. B. Sicherstellung der Rückverfolgbarkeit und Qualität der in der Produktion verwendeten Materialien), Qualitätssicherung, Qualitätskontrolle und vieles mehr, alles dokumentiert in einem Qualitätsmanagementsystem (QMS). GMP betont eine gründliche Dokumentation, die die Rückverfolgung jeder Aktivität während einer Herstellungscharge ermöglicht. (Für weitere Details zur Qualitätssicherung bietet dieses Paper einen lesbaren Überblick — es ist auf Zelltherapie fokussiert, aber die Prinzipien sind dieselben.)
Wie sieht GMP in der Praxis aus?
- Prozesse sind definiert und die Ausführung wird kontrolliert. Jeder Schritt im Herstellungsprozess und bei der Prüfung des Arzneimittels, das aus dem eigentlichen Wirkstoff (Drug Substance) und allen anderen Hilfsstoffen, Bindemitteln, Beschichtungen usw. besteht, muss durch ein Rezept/Verfahren definiert sein, dessen Ausführung aufgezeichnet und überprüft wird.
Dieses Rezept wird hochdetailliert sein: Es könnte beispielsweise drei verschiedene Schritte geben.
1 ml Reagenz A mit einem bestimmten Instrument und Modell abmessen.
-
Reagenz A mit Reagenz B in einem bestimmten Gefäß bestimmter Größe und Modells kombinieren.
-
Das Volumen der resultierenden Mischung mit einer in einer Standardarbeitsanweisung definierten Technik aufzeichnen.
-
Person A befolgt das Verfahren.
-
Nach jedem kleinen Schritt muss Person A, die ihn durchgeführt hat, neben diesem Schritt auf einem Blatt Papier (oder elektronischem Äquivalent) signieren und bestätigen, dass sie das Angegebene getan hat. Wenn ein Ergebnis aufzuzeichnen ist (z. B. wie in Schritt iii), schreibt sie dieses Ergebnis auf.
-
Person B beobachtet Person A bei der Durchführung des Schritts und signiert ebenfalls neben dem Schritt, um zu bestätigen, dass Person A ihn korrekt durchgeführt hat. Wenn Person A ein Ergebnis aufgezeichnet hat, bestätigt Person B die korrekte Aufzeichnung.
-
Person A muss für die Befolgung dieses Verfahrens geschult worden sein und dies nachweisen können. Dasselbe gilt für Person B, die Kontrollperson.
-
All dies wird für jeden “qualitätskritischen” Schritt durchgeführt.
-
Zwischenprodukte werden getestet und qualitätsgeprüft. Es gibt Tests von Zwischenprodukten während des gesamten Herstellungsprozesses, um sicherzustellen, dass sie innerhalb vordefinierter Spezifikationen liegen und z. B. keine Kontamination stattgefunden hat. Oft müssen neue Assays entwickelt werden, um zu testen, ob die erstellten Zwischenprodukte und der endgültige Wirkstoff den Spezifikationen entsprechen. Es reicht nicht aus, sich auf die Prüfung des Medikaments am Ende des Prozesses zu verlassen.
-
Aufzeichnungen werden von einer dedizierten Qualitätsfunktion geprüft und freigegeben. Bevor Patienten ein Medikament verwenden können, müssen alle Dokumente, die jeden Schritt des Prozesses aufzeichnen, von Mitarbeitern einer Abteilung überprüft werden, deren einzige Rolle die Qualitätssicherung ist.
Wenn die gesamte Herstellung abgeschlossen ist, haben Sie einen Satz von Dokumenten, die von Person A und B signiert wurden, wobei jedes einzelne einen Teil des gesamten Herstellungsprozesses darstellt.
-
Zusammen wird dieser Satz von Aufzeichnungen als Chargenprotokoll (Batch Record) bezeichnet. (Sie dokumentieren die Herstellung der Charge des Medikaments.)
-
Die Qualitätsfunktion (Qualitätssicherungsabteilung) der Produktionsstätte muss das Chargenprotokoll überprüfen.
GMP verlangt, dass die Qualitätsfunktion operativ unabhängig vom Team ist, das die Herstellung direkt durchführt, um Interessenkonflikte zu vermeiden. Die Qualitätsfunktion berichtet typischerweise an den CEO.
-
In einigen Ländern, z. B. in der EU, kann diese “Chargenfreigabe” nur von “Sachkundigen Personen” (Qualified Persons, QPs) durchgeführt werden — akkreditierte Personen mit spezifischer Expertise in der Arzneimittelherstellung.
-
Wenn ein Sponsor (das Unternehmen, dem das Medikament im Wesentlichen gehört — mehr dazu hier) die Herstellung auslagert, wird dieser Schritt sowohl vom Sponsor als auch vom Auftragnehmer durchgeführt, der die Herstellung direkt vornimmt.
-
Die Überprüfung umfasst beispielsweise die Kontrolle, ob das Produkt die vordefinierten Spezifikationen erfüllt und ob die Verfahren ordnungsgemäß befolgt wurden.
-
Dokumente werden für potenzielle Audits aufbewahrt. Die aus diesem Prozess resultierenden Dokumente und die Aufzeichnungen über die durchgeführte Überprüfung werden sicher aufbewahrt, versioniert und sind bei Audits für Regulierungsbehörden zugänglich.
Die Herstellung eines Medikaments bringt erheblichen Overhead mit sich. Kleine Mengen von Arzneimittelprototypen für sehr frühe Experimente können intern von Sponsoren synthetisiert werden, aber aufgrund all der oben aufgeführten Prozesse sind Kooperationen mit CDMOs oft notwendig — und diese haben üblicherweise Wartezeiten von 6-12 Monaten. Interessanterweise werden nur etwa 15 % des gesamten F&E-Budgets für die Herstellung aufgewendet, wobei der Großteil in Qualitätssicherung und Tests fließt. Die Kosten unterscheiden sich definitiv nach Therapiegebiet, aber ein GMP-Herstellungslauf erfordert oft eine Investition im ein- bis zweistelligen Millionenbereich, während Nicht-GMP-Chargen in der Regel deutlich unter einer Million Dollar liegen.
Arzneimittelentdeckung
Es gibt viele Wege, zu einem hypothetischen Kandidaten zu gelangen, von denen einige das In-silico-Screening von Tausenden von Verbindungen auf Wirksamkeit beinhalten, abhängig vom therapeutischen Ansatz (z. B. High Throughput Screening (HTS), andere sind einfache In-vitro-Reagenzglas- oder Zellkulturexperimente). Es gibt eine signifikante Reduktion der Vielzahl von Verbindungen während des Screenings — siehe die folgende Abbildung (Quelle) der Ausfallrate bei jedem Schritt der Arzneimittelentdeckung und -entwicklung.
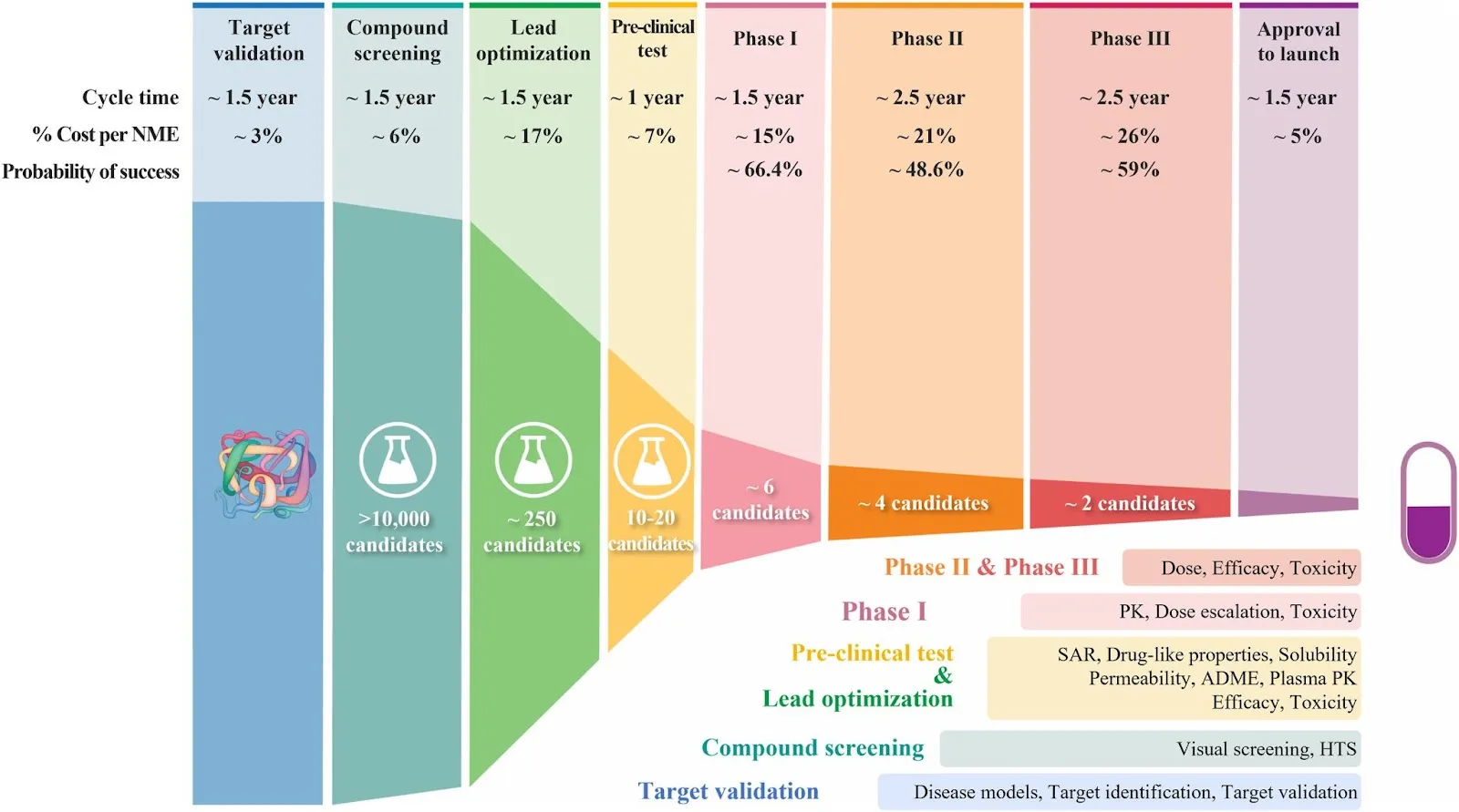
Obwohl es keine “Good Discovery Practices” gibt und keine formale behördliche Genehmigung erforderlich ist, unterliegen Wissenschaftler in Laboren stets Biosicherheitsstandards unter der Aufsicht von Behörden — staatliche Behörden für niedrigere Laborstufen (BSL-2 und darunter) und das CDC in den USA für höhere Laborstufen (BSL-3 und BSL-4). Zusätzliche Regulierung kann unter besonderen Umständen gelten, etwa bei Pathogenen besonderer Bedeutung (Federal Select Agent Program) oder aufgrund internationaler Zusammenarbeit.
Gescheiterte Kandidaten verbrauchen etwa ein Drittel der Gesamtausgaben eines Arzneimittelentwicklungsprogramms, d. h. Hunderte von Millionen Dollar, und erstrecken sich in der Regel über Jahre. “Aha-Momente”, bei denen Wissenschaftler erfolgreich Verbindungen identifizieren, die auf zentrale biologische Mechanismen abzielen, sind seltener als man hoffen würde.
Präklinische Forschung
Sobald ein Kandidat identifiziert ist, geht er in die präklinische Forschung. Diese Phase erforderte früher Tierversuche, aber die FDA kündigte 2022 an, dass sie nun auch Nicht-Tier-Organoid-Modelle akzeptiert.
In präklinischen Tierstudien werden verschiedene Parameter untersucht. Während die Details zur Durchführung der Prüfung von Arzneimittelsicherheit und -wirksamkeit an Tieren in GLP beschrieben sind, mit zusätzlichen Spezifikationen in verschiedenen regulatorischen Dokumenten wie der Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals oder den WHO-Leitlinien zur nichtklinischen Bewertung von Impfstoffen. Im Allgemeinen sind die folgenden Komponenten relevant:
-
Pharmakokinetik: Dieser Bereich untersucht die Absorption, Verteilung, den Metabolismus und die Ausscheidung des Arzneimittels, um sein Verhalten im biologischen System zu verstehen.
-
Toxikologie: Definiert dosisabhängige Auswirkungen auf verschiedene Organsysteme und Entwicklungsstadien von Organismen. Sie helfen, die sichere Startdosis in Studien am Menschen festzulegen, wobei die Dosen am No Observed Adverse Effect Level (NOAEL) bei Tieren mit Sicherheitsfaktoren multipliziert werden. (Eine detaillierte Erläuterung zur Extrapolation von Tier- auf Humandosen finden Sie hier.)
-
Pharmakodynamik: Bestimmt die Wirkungen des Arzneimittels auf den Körper, einschließlich des Wirkmechanismus und der Beziehung zwischen Arzneimittelkonzentration und Wirkung.
-
Wirksamkeitsstudien: Diese bestimmen, wie gut ein Arzneimittel seine beabsichtigte therapeutische Funktion erfüllt. Es umfasst Untersuchungen zu Dosis-Wirkungs-Beziehungen, um die optimale Dosierung für den gewünschten therapeutischen Effekt ohne schädliche Nebenwirkungen zu verstehen. Dies hilft, das therapeutische Fenster eines Arzneimittels zu definieren, ein kritischer Aspekt in der weiteren klinischen Entwicklung.
Einige dieser Tests können kombiniert werden; andere erfordern zusätzliche Studien. Beispielsweise testet eine einzelne Toxizitätsstudie üblicherweise ~100 Tiere von mindestens zwei Arten und kann auch verwendet werden, um zusätzliche pharmakologische Aspekte des Arzneimittels zu erfassen. Die Gesamtzahl und Art der Tiere kann weit darüber hinausgehen, wenn zusätzliche Tests in verschiedenen Modellorganismen erforderlich sind — abhängig von Faktoren wie der chemischen Zusammensetzung des Arzneimittels, dem Therapiegebiet, den Dosisstufen und den zu testenden Wirkungen.
Es gibt selten harte Anforderungen von Regulierungsbehörden bezüglich des spezifischen Tiermodells, aber es kann Erwartungen in Leitlinien geben, und es lohnt sich, mit erfahrenen Personen auf dem Gebiet zu sprechen (z. B. sind Syrische Hamster, obwohl nicht ausdrücklich in Leitlinien erwähnt, ein Standard für COVID-19-Impfstoffstudien gewesen, Schafe sind ein gängiger Organismus für Inhalationsstudien, und nichtmenschliche Primaten sind zunehmend gefragt für nahezu jede Art von Forschung, obwohl sie von Regulierungsbehörden oft aktiv abgeraten werden).
Überraschenderweise sind die Kostentreiber in präklinischen Studien nicht die Anzahl der Tiere, sondern das Arzneimittel selbst, die Personalkosten beim Studienanbieter (Anzahl der Dosen, Blutentnahmen usw.) und Analyse-/Pathologiekosten. In aktuellen US-Industrieangeboten für Studien mit ~100 Mäusen mit mehreren Injektionen und Blutentnahmen über 35 Tage lagen die Kosten pro Tier bei etwa 750 Dollar, insgesamt 75.000 Dollar. Die Kosten für die Herstellung des notwendigen Nicht-GMP-Arzneimittels können dies leicht verdoppeln, wobei die Analysekosten das 0,5- bis 1,5-Fache der Studienkosten betragen. Diese Zahlen können natürlich stark variieren, abhängig vom konkreten Tierstudienplan, der typischerweise vom Studienanbieter bereitgestellt wird, sobald alle relevanten Variablen bekannt sind. Die präklinische Forschung dauert etwa 31 Monate und macht etwa 43 % der gesamten F&E-Ausgaben eines Arzneimittelentwicklungsprogramms aus.
Klinische Entwicklung
Sobald ein potenzielles Arzneimittel die präklinischen Studien besteht, geht es in klinische Studien am Menschen über. Die klinische Entwicklung ist in Phase-I-, -II-, -III- und -IV-Studien unterteilt, jeweils mit unterschiedlichen Zielen und zunehmenden Umfängen. Eine einzelne Arzneimittelzulassung umfasst üblicherweise mehrere Studien über verschiedene Phasen hinweg, die in verschiedenen Patientensubgruppen, Dosisstufen oder Endpunkten testen. In der EU werden jährlich ~3.700 klinische Studien genehmigt, was 46 % von fast 8.000 Anträgen ausmacht.
Als grobe Orientierung (verschiedene Quellen zusammenfassend hier, hier und hier):
-
Phase I: Umfasst bis zu 100 gesunde Teilnehmer; ~2 Studien pro Entwicklungsprogramm
-
Phase II: Umfasst ~100-500 Patienten und zielt darauf ab, einen Wirksamkeitsnachweis zu erbringen; ~2 Studien pro Entwicklungsprogramm
-
Phase III: Getestet an einer Gruppe von ~500-5000+ Patienten, um weitere Sicherheits- und Wirksamkeitsdaten zu generieren, die ausreichen, um Regulierungsbehörden von einer Zulassung zu überzeugen; ~3 Studien pro Entwicklungsprogramm
-
Phase IV: ~100-1500+ Patienten, abhängig von Studie und untersuchtem Arzneimittel; ~3 Studien pro Entwicklungsprogramm.
Die Gesamtteilnehmerzahl eines klinischen Entwicklungsprogramms kann von Hunderten bis über 30.000 vor der vollständigen Arzneimittelzulassung reichen. Beachten Sie, dass bei bestimmten Indikationen, insbesondere selteneren Erkrankungen, die Patientenzahl in jeder Phase deutlich kleiner sein kann als angegeben. Die Abbildung (Quelle) zeigt die durchschnittlichen Kosten pro Studie (beachten Sie, dass es wesentlich teurere Ausreißer gibt).
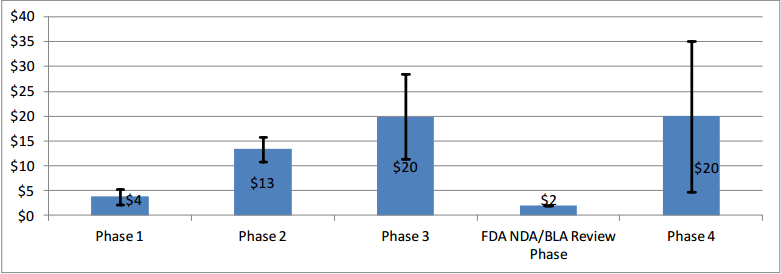
Klinische Tests dauern etwa doppelt so lang wie die Entdeckungs- und präklinische Phase zusammen. Die mediane Dauer nicht-onkologischer Arzneimittel in klinischen Studien liegt bei etwa 5,9 bis 7,2 Jahren. Bei Onkologiemedikamenten verlängert sich dieser Zeitraum auf durchschnittlich 13,1 Jahre. Aufgeschlüsselt nach klinischer Entwicklungsphase, erwarten Sie ~1,6, 2,9 und 3,8 Jahre in Phase I, II bzw. III.
Die beispiellose Geschwindigkeit der COVID-19-Impfstoffentwicklung weckte bei vielen die Hoffnung auf kürzere Zeitrahmen in der Zukunft. Es besteht oft der Eindruck, dass die langen Zeiträume auf langsame Pharmaunternehmen und regulatorische Hürden zurückzuführen sind. Sponsoren machen Fehler wie schlechtes Studiendesign, ineffektive Standortwahl, mangelhafte Rekrutierung, Patientenbelastung/Sicherheitsprobleme und fehlerhafte Studiendurchführung, die Verzögerungen oder sogar das Scheitern verursachen. Es ist jedoch wichtig zu beachten, dass einige Faktoren wirklich schwer zu beschleunigen sind. Die Rekrutierung bestimmter Teilnehmertypen (z. B. bei seltenen Erkrankungen) kann sehr herausfordernd sein. Während Surrogat-Blutmarker leicht zu erheben sind, erfordern die Endpunkte, die uns am meisten interessieren — Mortalität und Morbidität — längere Beobachtungszeiträume. Zum Beispiel könnte man fragen: “Sterben weniger Menschen aufgrund kardiovaskulärer Vorteile über ein, drei oder bis zu fünf Jahre?” — dies wäre grundsätzlich unmöglich zu beschleunigen.
GCP ist die wichtigste einzelne Leitlinie für die Durchführung klinischer Studien. Sie deckt ethische Erwägungen, informierte Einwilligung, Rollen mit ihren Verantwortlichkeiten, Qualitätssicherungs- und Datenmanagementprotokolle und vieles mehr ab. Die Einhaltung von und Schulung für GCP ist an Prüfzentren klinischer Studien obligatorisch, und Hauptprüfer können persönlich für Verstöße haftbar gemacht werden. Eine klare und umfassende Erläuterung, wie alle diese Anforderungen erfüllt werden, muss bei Regulierungsbehörden eingereicht werden (in den USA auch als Investigational New Drug (IND) Submission bekannt) sowie bei lokalen IRBs. Drei der relevantesten Dokumente sind:
-
Das Studienprotokoll enthält alle primären/sekundären/explorativen Endpunkte, den statistischen Analyseplan und andere relevante Beschreibungen, z. B. Randomisierungsstrategien. Die klinische Studie muss gemäß Protokoll durchgeführt werden. Abweichungen vom Protokoll müssen aufgezeichnet werden und können die Validität der Studie gefährden, wenn sie nicht richtig gehandhabt werden. Viele Studienprotokolle werden veröffentlicht und sind entweder über Clinicaltrials.gov oder über die Supplemente der Publikationen der jeweiligen Studie zugänglich.
-
Die Prüferinformation (Investigator’s Brochure, IB) richtet sich an die Ausbildung von Ärzten für die Durchführung der klinischen Studie und die Möglichkeit, sich selbst, das Personal und Patienten über das untersuchte Produkt zu informieren. Dies umfasst Informationen zur Zusammensetzung und Herstellung des Arzneimittels, nichtklinische Studienergebnisse (üblicherweise aus In-vitro- und In-vivo-Tests), die das Sicherheitsprofil des Arzneimittels demonstrieren und Studien am Menschen rechtfertigen. Die GCP-Leitlinie beschreibt in ihrem letzten Kapitel, welche Abschnitte in einer IB enthalten sein sollten. Obwohl seltener öffentlich verfügbar, ist ein gutes Beispiel die Prüferinformation zum mRNA-COVID-19-Impfstoff von Pfizer.
-
Patienteneinwilligung (Informed Consent) IRBs bewerten, dass die Rechte und das Wohlergehen der menschlichen Teilnehmer geschützt sind, der potenzielle Nutzen der Forschung die möglichen Risiken überwiegt und die Untersuchung ethisch und in Übereinstimmung mit anerkannten Vorschriften durchgeführt wird. IRBs stellen oft Checklisten oder Vorlagen bereit, welche Informationen und Formatierungen sie erwarten. CROs kennen häufig besondere Präferenzen der IRBs und können entsprechend beraten.
Sowohl die Regulierungsbehörde als auch das IRB bewerten das Dokumentenpaket nach Eingang, um zu beurteilen, ob die vorgeschlagenen Studien sicher durchführbar sind. Es kann sehr unterschiedliche Zeitrahmen über verschiedene Rechtsordnungen/IRBs geben, und fachkundige regulatorische Beratung kann manchmal Monate einsparen. Die Genehmigung für eine Phase-1-Studie in Australien beispielsweise kann in wenigen Wochen erteilt werden, während die USA oder die EU leicht 6 Monate dauern können.
Ähnlich wie im GMP-Abschnitt bietet das Folgende ein konkretes Beispiel aller Aktivitäten in einer klinischen Studie, die ein Sponsor im Blick behalten muss:
- Studienbetrieb
CRO- und Standortauswahl (in Abstimmung mit Akteuren — klinisch, regulatorisch)
-
Zusammenstellung und Einreichung von Dokumenten für klinische Studienanträge (CTAs)
-
Zusammenstellung und Einreichung von Dokumenten für Ethikkommissionen
-
Studienabschlüsse
-
Projektmanagementpläne für die Studien, inkl. Zeitplanmanagement
-
Übergeordnetes Projektmanagement der klinischen Studie
-
Management von Dienstleistern, die nicht anderen Fachexperten zugeordnet sind
-
Studienversicherungen
-
Rechtliche Vereinbarungen bezüglich der Studie und Studiendienstleister
-
Risikolog
-
Maßnahmenlog
-
Identifikation und kontinuierliche Zusammenarbeit mit Prüfzentren und CROs
-
Koordination von Standortbesuchen in Zusammenarbeit mit relevanten Fachexperten
-
Studien-Lieferkette
Transport des Prüfpräparats (IMP) zu den Prüfzentren
-
Beschaffung von Placebos / Vergleichspräparaten
-
Management und Überwachung von Depots vor/während/nach der Studie
-
Sicherstellung einer adäquaten Beschaffung von Nicht-IMP-Materialien, entweder direkt oder in Zusammenarbeit mit den jeweiligen Fachexperten
-
Datenmanagement / -verarbeitung / -analyse
Überwachung der medizinischen Monitore
-
IRT-Überwachung und -Management
-
EDC-Einrichtung, Überwachung, Management und Aufsicht
-
Datenbereinigung
-
Statistische Analyse
-
Medizinisches Schreiben
Entwicklung, Überarbeitung und Pflege von
Protokollen
-
IBs
-
Einwilligungserklärungen (ICFs) und anderen patientengerichteten Materialien
-
Laborhandbüchern und Apothekenhandbüchern
-
relevanten zusätzlichen Materialien, die für Studienanträge und -durchführung erforderlich sind
-
Entsprechende Übersetzungen von Materialien, wenn erforderlich
-
Veröffentlichung von Studienergebnissen
-
Teilnehmerwohl / Pharmakovigilanz
Risikoanalyse aller Sicherheitsbedenken, die während der Studie für Teilnehmer auftreten könnten
-
Entwicklung, Überarbeitung und Pflege von SMP / MMP
-
Einrichtung und Überwachung von Sicherheitsdatenbanken
-
Erstellung von Sicherheitsberichten
-
Management von DSMBs / IMMs
-
Überprüfung und kontinuierliche Überwachung jeglicher Art von unerwünschten Ereignissen in klinischen Studien
-
Studientests
Identifikation fähiger Dienstleister, die die für unsere Studie notwendigen Tests durchführen können.
-
Definition der notwendigen Tests, einschließlich körperlicher Untersuchung, Immunogenitäts-/Sicherheitslabore, genetischer Tests usw.
-
Definition der Materialien / Verfahren, die für angemessene Tests benötigt werden (z. B. Röhrchen, Tupfer, Messgeräte usw.)
-
Überprüfung der Validität der von Testdienstleistern gelieferten Daten
-
Abteilungsübergreifende Schnittstellen
Enge Zusammenarbeit mit anderen Abteilungen im gesamten Unternehmen (z. B. Herstellung, präklinische Wissenschaft, Betrieb usw.)
- Studien-Qualitätssicherung & Monitoring
Aufsicht über die Entwicklung relevanter SOPs und Arbeitsanweisungen in Zusammenarbeit mit dem QA-Team
-
Überprüfung der laufenden Verwendung von SOPs / Arbeitsanweisungen im gesamten Team
-
Regulatorische Compliance
Kontinuierliche gegenseitige Aktualisierung der regulatorischen Teams sowie der Geschäftsführung
-
Zusammenarbeit bei der Definition von Fahrplänen für die Trajektorie des Studienteams zusammen mit regulatorischen Teams und der Geschäftsführung
-
Information des restlichen Teams über Fortschritte zwischen den jeweiligen Parteien
Die obige Liste veranschaulicht, dass Sie ein interdisziplinäres Team benötigen. Bei vielen Unternehmen sind mehrere dieser interdisziplinären Teams aufeinander geschichtet:
-
Globale Entwicklungsteams denken auf der Ebene eines Therapiegebiets (etwa “Welchen Impfstofftyp oder welches Blutdruckmedikament sollten wir weiterentwickeln?”)
-
Klinische Programmteams konzentrieren sich auf die Evaluation von Arzneimittelkandidaten (“Wie sammeln wir alle notwendigen Daten für die Zulassung dieses Arzneimittelkandidaten?”)
-
Klinische Studienteams führen einzelne klinische Studien durch (“Wie führen wir die Studie effizient und mit den wertvollsten Daten durch?”)
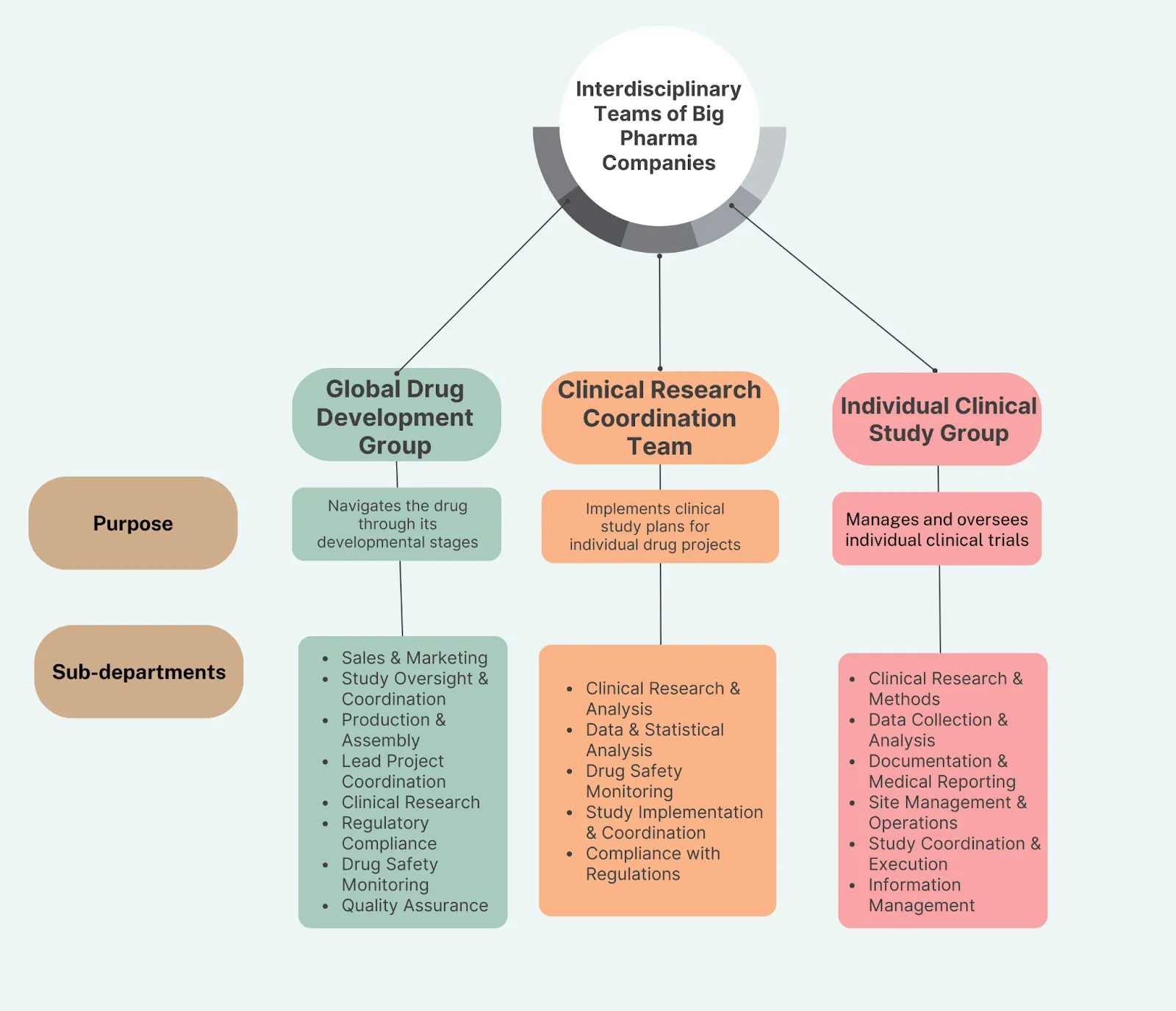
Marktzulassung
Wenn ein Arzneimittel alle diese Phasen mit guten Ergebnissen durchläuft, wird ein Antrag auf Marktzulassung eingereicht. In den USA bedeutet dies die Einreichung eines New Drug Application (NDA) oder Biologics License Application (BLA). Ähnliche Zulassungsanträge in anderen Ländern sind typischerweise in drei Hauptteile gegliedert: präklinische Daten, detaillierte Herstellungsmethodik zur Sicherstellung der Reproduzierbarkeit und natürlich Informationen zu klinischen Studien. Jeder Abschnitt ist ungefähr gleich lang und trägt zu einem “Dossier” bei, das Tausende von Seiten umfasst. Die FDA prüft alle Daten gründlich und entscheidet, ob das Arzneimittel zugelassen wird. Diese Gründlichkeit wird durch dieses umfangreiche Inhaltsverzeichnis über zehn Seiten im NDA veranschaulicht.
Jedes Jahr bearbeitet die FDA ~400 originale Investigational New Drug-Anträge, wobei von 2010 bis 2019 durchschnittlich 38 neue Medikamente pro Jahr zugelassen wurden. Davon werden 3-4 % später zurückgezogen. Weltweit liegt die Rückzugsrate nach der Zulassung bei etwa 10 %. Jede Änderung des Herstellungsprozesses, der Indikationen oder der Dosierung erfordert zusätzliche Prüfung und erneute Genehmigung. Die Wahrscheinlichkeit, dass ein Arzneimittelentwicklungsprogramm eine Zulassung erhält, liegt bei eins zu sieben, wobei die Raten von 3,4 % für Onkologie bis maximal 33,4 % für Impfstoffe reichen.
Post-Marketing-Überwachung
Der Arzneimittelentwickler wird von den Regulierungsbehörden erwartet, die Arzneimittelsicherheit weiterhin zu überwachen, sobald es in den öffentlichen Gebrauch gelangt. Dies umfasst die Überprüfung von Problemberichten, die Regulierung von Werbung für verschreibungspflichtige Medikamente, die Durchführung routinemäßiger Inspektionen usw. Alle Daten werden in Post-Marketing-Überwachungsberichten zusammengefasst, die vom Sponsor eingereicht werden, zunächst in Sechs-Monats-Intervallen, die sich im Laufe der Zeit auf drei oder mehr Jahre verlängern. Die FDA kann auch weitere Wirksamkeitsstudien verlangen und führt selbst Audits durch, um die fortlaufende Einhaltung und Arzneimittelsicherheit sicherzustellen. Sowohl Ärzte als auch Patienten werden ermutigt, alle Nebenwirkungen, die sie bei einem Medikament bemerken, den Regulierungsbehörden zu melden — siehe hier für die Kontaktdaten der FDA und der EMA.
In unserem nächsten Beitrag werden wir die Ursprünge der Arzneimittelregulierung betrachten und untersuchen, wie historische Ereignisse und wichtige regulatorische Meilensteine zu den heutigen strengen Praktiken in der Arzneimittelentwicklung beigetragen haben. Das Verständnis der Entwicklung der Arzneimittelregulierung wird einen Kontext zu den bestehenden rigorosen Prozessen liefern und zeigen, wie sie die pharmazeutische Landschaft weiterhin prägen.