Ursprünge der Arzneimittelregulierung (3/6 Artikelserie zur Arzneimittelentwicklung)
In unseren letzten Beiträgen dieser Serie haben wir die beteiligten Akteure und die notwendigen Schritte für die Markteinführung eines Medikaments betrachtet. Man fragt sich unweigerlich — warum erfordert die Arzneimittelentwicklung all diese Strenge, Zeit und Kosten? Das Verständnis der Geschichte der Arzneimittelentwicklung kann Kontext und Weitsicht bieten, um ihre aktuelle und zukünftige Richtung zu erkennen. Wenn Sie die Geschichte betrachten, werden Sie feststellen, dass Veränderungen oft auf Reaktionen auf Tragödien zurückgeführt werden, obwohl in vielen Fällen eine genauere Betrachtung zeigt, dass Reformen bereits im Gange waren.
Wir werden auch sehen, dass die regulatorischen Geschichten verschiedener Länder begannen sich zu überschneiden, als internationale Bemühungen zur Harmonisierung von Gesetzen erfolgreich wurden, insbesondere in der zweiten Hälfte des 20. Jahrhunderts. Beachten Sie, dass die nachfolgende Perspektive sich auf einen Überblick hauptsächlich aus US-amerikanischer Sicht konzentriert — wir ermutigen Leser, Dan Carpenters historische Analyse der FDA für ein detaillierteres Verständnis zu konsultieren und andere Autoren für die jeweiligen Rechtsordnungen heranzuziehen.
USA: Die Ursprünge der Arzneimittelregulierung nachverfolgen
Wir wussten es noch nicht, aber der Kauf von Medikamenten in den 1900er Jahren war wie ein Würfelspiel. Zu dieser Zeit behaupteten viele “Patentmedizinen”, alles heilen zu können, von Kopfschmerzen bis Liebeskummer. Zum Beispiel wurde Dr. D. Jaynes Tonic Vermifuge 1896 als Allheilmittel für Hals- und Atemwegserkrankungen und als Wurmmittel beworben.

Eine Werbung für Dr. D. Jaynes Tonic Vermifuge
Eine weitere ikonische Darstellung von Patentmedizinen im 19. Jahrhundert war “Schlangenöl”, das für seine angeblichen Heilkräfte angepriesen wurde, von der Heilung von Kopfschmerzen bis zur Linderung chronischer Schmerzen. Die Legende des Schlangenöls symbolisiert die archetypische Patentmedizin und verkörpert die unkontrollierten Behauptungen und Placebomittel, die in einer Ära vor strenger Arzneimittelregulierung und -prüfung allgegenwärtig waren.

Ein Foto von berühmten “Schlangenliniment”-Flyern und -Flaschen von Clark Stanley in den 1800er Jahren
Dies war die Landschaft vor der Arzneimittelregulierung. Stellen Sie sich vor, Sie sind ein Wissenschaftler in diesem Szenario, begierig darauf, Ihr neues Medikament zu testen und zu vermarkten. Vielleicht hätte sich dieser Mangel an Regulierung befreiend angefühlt, aber im Laufe der Zeit erkennen Sie, wie der entscheidende Bedarf an Standardisierung und Sicherheitsprotokollen offensichtlich wird. Mit der Zeit werden sich weiterentwickelnde regulatorische Rahmenbedingungen Ihre Arbeit zunehmend prägen.
Seit den 1880er Jahren gab es Bemühungen, die ordnungsgemäße Kennzeichnung von Lebensmitteln und Medikamenten zu regulieren und Verunreinigungen in beiden Kategorien zu verhindern. Schäden durch unregulierte Lebensmittel und Arzneimittel, wie der kontaminierte Diphtherie-Antitoxin-Vorfall, waren weit verbreitet und führten zum Tod mehrerer Kinder. Erstmals institutionalisierte der Biologics Control Act (1902) Inspektionen und Reinheitsprüfungen in den USA und markierte einen Schritt in Richtung formaler Regulierung zur Sicherstellung der Arzneimittelsicherheit und -wirksamkeit. Kurz darauf leistete die American Medical Association (AMA) Pionierarbeit in der Selbstregulierung mit einem freiwilligen Arzneimittelzulassungsprogramm, bei dem Unternehmen Wirksamkeitsnachweise für ihre Medikamente vorlegen mussten, bevor sie in AMA-Zeitschriften werben durften. Diese schufen Präzedenzfälle für weitere regulatorische Maßnahmen wie den Pure Food and Drug Act (1906) für die US Food and Drug Administration (FDA), der den Verkauf kontaminierter Lebensmittel oder Fleisch illegal machte. Das Gesetz verlangte wahrheitsgemäße Kennzeichnung — niemand durfte mehr “das Blaue vom Himmel” auf ein Etikett versprechen — zumindest war das das Ziel.
USA: Reaktivität, Proaktivität und regulatorische Maßnahmen
Viele Regulierungen resultierten aus dem öffentlichen Aufschrei, der auf Gesundheitskatastrophen folgte.
-
Federal Food, Drug, and Cosmetic Act (1938): Forderte den Sicherheitsnachweis vor der Zulassung, eingeführt nach einer verheerenden Tragödie mit einem schlecht formulierten Antibiotikum, die zu über hundert Todesfällen führte.
-
Good Manufacturing Practices (GMP, Anfang der 1940er): Entwickelt nach einem Vorfall mit einer Charge von Sulfonamiden, die mit dem Beruhigungsmittel Phenobarbital kontaminiert waren (mit Hunderten von Toten und Verletzten).
-
Nürnberger Kodex von 1947: Diente als Leitlinie für klinische Studien (mit der Festlegung von Prinzipien für ethische Forschung wie freiwillige Einwilligung) im Gefolge unmenschlicher Experimente der Nazis in Deutschland, aufbauend auf der Deklaration von Helsinki von 1964.
-
FDAs Verbot von GAS-Impfstoffen (1979): Ein wohl überreaktives Verbot der Verwendung von Gruppe-A-Streptokokken (GAS)-Organismen und ihren Derivaten in Impfstoffen, nachdem zwei oder drei von 21 gesunden Probanden in einer Studie 1969 nach der Impfung rheumatisches Fieber entwickelt hatten. Trotz der langen Geschichte erfolgreicher M-Protein-Impfstoffe — die auf Komponenten von Bakterien wie GAS abzielen — hat das FDA-Verbot die Forschung in diesem Bereich fast drei Jahrzehnte lang effektiv unterdrückt. Die Entscheidung wurde angesichts des hohen ungedeckten medizinischen Bedarfs im Zusammenhang mit GAS als besonders bedeutsam erachtet. Erst 2006 wurde das Verbot aufgehoben und die Wiederaufnahme wichtiger Forschung und potenzieller Durchbrüche ermöglicht.
-
Schnellerer Zugang zu lebensrettenden Medikamenten (1990er Jahre): Wenn auch sorgfältig überwacht, Zugang zu lebensrettenden Medikamenten, nachdem AIDS-Aktivisten die FDA kritisiert hatten, den Zugang zu potenziell lebensrettenden Medikamenten zu blockieren und langsame Arzneimittelzulassungen während der 1990er Jahre.
-
Die FDA Gender-Richtlinie (1993): Verlangte die Prüfung von Medikamenten an beiden Geschlechtern und hob eine Richtlinie von 1977 auf, die fruchtbare Frauen von frühen klinischen Studien ausschloss (was eine Reaktion auf die Thalidomid-Katastrophe war, ein Ereignis, das weiter unten beschrieben wird).
Es ist jedoch wichtig anzumerken, dass es verlockend ist zu denken, die Arzneimittelregulierung sei rein aus öffentlichem Aufschrei entstanden, eine Art legislative Reflexreaktion auf aufsehenerregende Katastrophen, aber das ist nur ein Teil der Geschichte. Viele Reformen schwelten bereits, bevor eine Tragödie die Dringlichkeit erhöhte. Auch die politische Entwicklung hin zu einer proaktiven statt reaktiven Regulierung ist bemerkenswert. Waren gesetzgeberische Änderungen früher eher krisengetrieben, gibt es heute ein erkennbares Bemühen, Risiken zu antizipieren und präventive Maßnahmen umzusetzen. Freiwillige Leitlinien haben ebenfalls eine Rolle gespielt, die manchmal aus bewährten Industriepraktiken entstanden und später zu regulatorischen Erwartungen wurden.
-
Bereits 1933 — um die Mängel des Pure Food and Drug Act von 1906 aufzuzeigen und die Notwendigkeit eines neuen Gesetzes zu veranschaulichen — startete die FDA die “American Chamber of Horrors”, eine Ausstellung mit problematischen Produkten wie unsicheren Medizinprodukten (z. B. einem schädlichen Gebärmutterstützer und einem tödlichen Diätmedikament) und giftigen Kosmetika. First Lady Eleanor Roosevelt, die die Ausstellung besichtigt hatte, nutzte sie, um stärkeren Verbraucherschutz voranzutreiben. Dies war ein präzises Beispiel dafür, dass die FDA die Initiative ergriff, um gesetzgeberische Veränderungen voranzutreiben.
-
FDA “Chargen-Zertifizierung”** (Anfang der 1940er):** Vorgeschrieben für bestimmte Schlüsselmedikamente, beginnend mit Insulin 1941 und dann Penicillin 1945. Unternehmen mussten Proben jeder Charge zur FDA-Prüfung einreichen, bevor sie auf den Markt gebracht werden konnten. Die Regelung wurde später auf alle Antibiotika ausgeweitet, aber 1983 eingestellt — ein seltenes Beispiel dafür, dass Vorschriften weniger streng werden.
-
Der Belmont Report (1979): Legte ethische Standards für die Forschung mit menschlichen Probanden fest und betonte Respekt vor Personen, Wohltätigkeit und Gerechtigkeit. Wenn Sie der Wissenschaftler sind, läge die Verantwortung nun bei Ihnen sicherzustellen, dass jeder Schritt der Arzneimittelentdeckung und klinischen Prüfung diesen ethischen Leitlinien entspricht — ein Zeugnis für die sich entwickelnde Integrität des Fachgebiets. Er hatte tiefgreifende Auswirkungen auf die Vorschriften für klinische Forschung in den USA und leitete ethische Erwägungen in medizinischen Studien. Im gleichen Zeitraum machten auch Bioethik-Komitees in anderen Ländern Fortschritte, veröffentlichten vergleichbare Leitlinien und stärkten ein globales Bekenntnis zu ethischen Standards.
Die Thalidomid-Katastrophe (späte 1950er — frühe 1960er Jahre) ist ein bedeutendes Kapitel in der Geschichte der Arzneimittelentwicklung und verdeutlicht die historische Spannung zwischen Reaktivität und Proaktivität. Thalidomid — ein Medikament, das ursprünglich zur Behandlung von Schwangerschaftsübelkeit bei schwangeren Frauen zugelassen und in den 1950er Jahren häufig verwendet wurde — führte zu schweren angeborenen Behinderungen, die in den 1960er Jahren sichtbar wurden, und entfachte den öffentlichen Ruf nach strengeren Arzneimittelgesetzen. US-Senator Kefauver, der sich bereits für Arzneimittelreformen einsetzte, nutzte diese Krise, um einen ins Stocken geratenen Gesetzentwurf im Kongress wiederzubeleben. Im Juli 1962 machte Kefauver die schädlichen Wirkungen des Medikaments publik und gab seinem Gesetzentwurf, der seiner Meinung nach an Schwung verloren hatte, neues Leben.
Abgesehen von den zahlreichen Reaktionen katalysierte die Tragödie umfassende Reformen in der Arzneimittelregulierung in den USA und weltweit. In den USA führte die Katastrophe zu der Vorschrift, dass jeder Sponsor, der eine klinische Untersuchung eines Medikaments plant, der FDA einen detaillierten Studienplan vorlegen und dessen Wirksamkeit nachweisen muss, nicht nur dessen Sicherheit. International trieb das Ereignis zentralisierte Zulassungsverfahren in der EU für neue Arzneimittelbewertungen voran. Zum Beispiel verabschiedete das Vereinigte Königreich den Medicines Act von 1968, der umfassende Arzneimittelklassifikationen einführte. Allgemein diente die Thalidomid-Katastrophe als Weckruf für die Branche und katalysierte Reformen, die nicht nur reaktiv, sondern auch umfassend und robust waren.
Globale Harmonisierung für die Arzneimittelentwicklung
Die International Conference on Harmonisation (ICH) zielte darauf ab, den Arzneimittelentwicklungsprozess ab den 1990er Jahren zu standardisieren (ähnlich wie USB-C versucht, Anschlüsse zu standardisieren). Die ICH-Initiative stellt einen kollektiven Schritt zu einem strafferen und einheitlicheren globalen Arzneimittelentwicklungsparadigma dar und leitet eine neue Ära kooperativer Bemühungen unter Arzneimittelherstellern über Grenzen hinweg ein. Durch die Zusammenführung von Regulierungsbehörden aus Europa, Japan und den USA stellt die ICH sicher, dass ein in einer Region getestetes und zugelassenes Medikament leichter Akzeptanz in anderen finden kann, was den globalen Zugang zu wichtigen Medikamenten beschleunigt.
Von ihrer Website: “Die Mission der ICH ist es, weltweit eine größere Harmonisierung zu erreichen, um sicherzustellen, dass sichere, wirksame und qualitativ hochwertige Arzneimittel auf die ressourceneffizienteste Weise entwickelt und registriert werden. Die Harmonisierung wird durch die Entwicklung von ICH-Leitlinien in einem Prozess wissenschaftlichen Konsenses erreicht, bei dem Experten aus Regulierung und Industrie Seite an Seite arbeiten.”
Die ICH hat umfangreiche Prozesse für neue Harmonisierungen, Klarstellungen, Revisionen und Pflege entwickelt und verfügt über ~150 Leitliniendokumente zu Sicherheit, Wirksamkeit, Qualität und Sonstigem. In Schritt 5 des ICH-Prozesses werden harmonisierte ICH-Leitlinien von ICH-Regulierungsmitgliedern und -Beobachtern in ihrem jeweiligen Land/ihrer jeweiligen Region umgesetzt. Die Umsetzung und Einhaltung der ICH-Leitlinien innerhalb der Länder/Regionen der Regulierungsmitglieder und -Beobachter werden durch unabhängige Drittanbieter-Umfragen überwacht (siehe Projektbericht 2021). Die ICH hat insgesamt 21 Mitgliedsorganisationen, jede mit zwei Vertretern. Viele Regulierungsbehörden integrieren ICH-Leitlinien in nationales Recht, wodurch die Einhaltung zu einer gesetzlichen Anforderung wird.
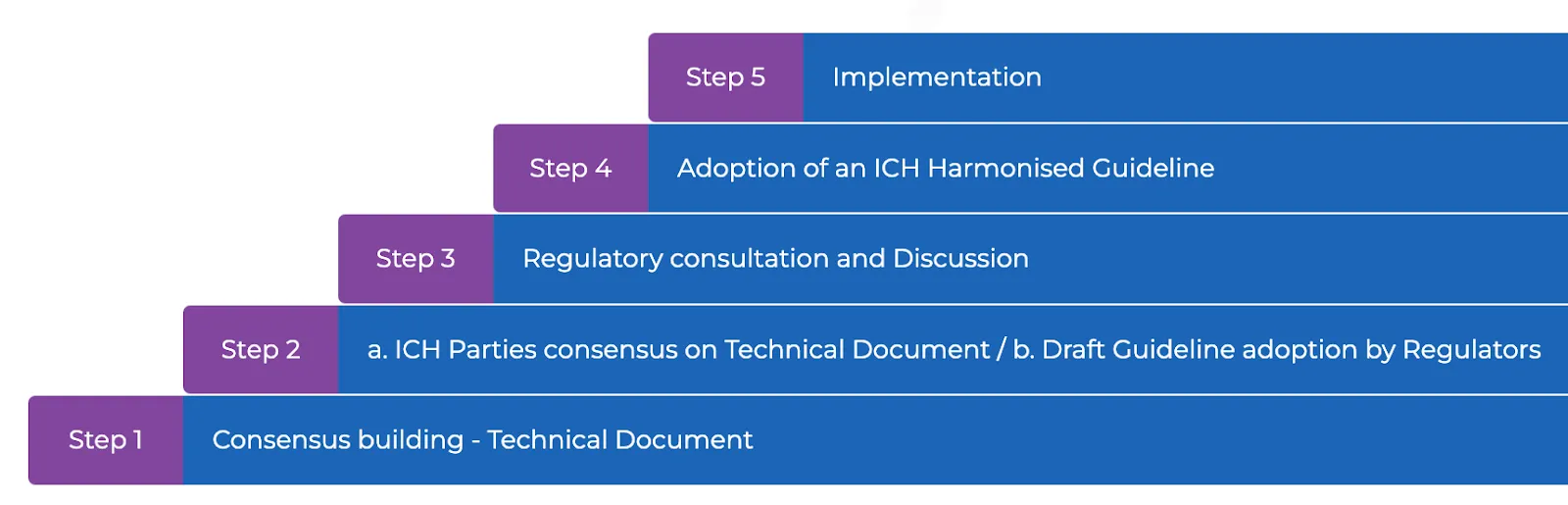
Der ICH-Prozess.
Ein Beispiel für den ICH-Prozess in Aktion: Im Jahr 2023 befinden sich die Good Clinical Practice (GCP)-Leitlinien der ICH in laufender Überarbeitung. Sie wurden 1996 initiiert, zuletzt 2016 aktualisiert, und ihre neueste Revision ist derzeit als Entwurf zugänglich. Diese Leitlinien entwickeln sich weiter und konzentrieren sich auf neue Studientypen und Datenquellen, was das Engagement von Regulierungsbehörden und Industrie widerspiegelt, der Entwicklung voraus zu sein.
Globale Meilensteine der Regulierung: Eine Tour von Peking bis Russland
Während die Geschichte in vielen westlichen Ländern ungefähr ähnlich wie in den USA verläuft, bietet dieser Abschnitt einen sehr kurzen Blick über Grenzen und Ozeane hinweg; die Geschichte der Arzneimittelregulierung entfaltet sich auch mit eigenständigen Kapiteln, die in verschiedenen Nationen geschrieben wurden. Jedes Land hat seine historischen Ereignisse und Reformen, die seine regulatorische Landschaft formen. Dies soll einen kompakten Überblick bieten und den Blick von einer US-zentrierten Sicht auf die Arzneimittelentwicklung weglenken — wir haben Quellen für tiefere Einblicke in spezifische Länder verlinkt und beanspruchen keineswegs, signifikante Expertise für eine dieser Regionen zu haben. Lassen Sie uns einige wichtige Meilensteine in bestimmten Ländern erkunden.
Brasilien
-
Die Nationale Gesundheitsüberwachungsbehörde oder ANVISA (1999): Gegründet zum Schutz der Gesundheit der Bevölkerung durch sanitäre Kontrolle.
-
Gesetz Nr. 13.411 (2016): Veränderte das pharmazeutische regulatorische Umfeld, senkte bürokratische Hürden in der klinischen Forschung und verbesserte die pharmazeutische Registrierung und Post-Marketing-Überwachungsprozesse.
-
Weiterlesen: Arzneimittelregistrierung in Brasilien
China
-
The Pharmaceutical Company of China (1950): Beauftragt mit der Übernahme des nationalen Großhandels mit Arzneimitteln, zu einer Zeit, als Krankenhäuser/Kliniken vom privaten Sektor in direkte staatliche Kontrolle überführt wurden.
-
GCP-Einführung durch die State Drug Administration oder SDA (2001)
-
CFDA-Verbesserungen (2015): Von der China Food and Medication Administration angekündigt, um die Arzneimittelzulassungsprozesse zu beschleunigen und chinesischen Patienten schnelleren Zugang zu neuen Medikamenten zu geben.
-
ICH-Mitgliedschaft (2018): China wurde Vollmitglied der ICH und demonstrierte damit sein Engagement für die Harmonisierung seiner pharmazeutischen Regeln mit globalen Normen (und die Reichweite der ICH).
-
Weiterlesen: Chinas Geschichte der Arzneimittelregulierung
Indien
- Vor der Regulierung
Praxis traditioneller indischer Medizinsysteme (Vor dem 20. Jahrhundert): Verwendung natürlicher Inhaltsstoffe wie Ayurveda, Siddha und Unani zur Behandlung.
-
Importära (20. Jahrhundert): Die meisten Medikamente wurden während der Kolonialzeit aus Europa importiert.
-
Die frühe pharmazeutische Industrie in Indien begann mit einigen wenigen einheimischen Unternehmen, die zu Beginn des 20. Jahrhunderts grundlegende Medikamente herstellten und die lokale pharmazeutische Entwicklung einschränkten.
-
Drugs and Cosmetics Act (1940): Etablierte die regulatorische Kontrolle über Import, Herstellung, Vertrieb und Verkauf von Arzneimitteln und Kosmetika in Indien und machte den Verkauf minderwertiger Medikamente zu einem schweren Vergehen.
1945 erließ die Regierung die Drugs and Cosmetics Rules, um Arzneimittel in bestimmte “Kategorien” (Schedules) einzuteilen und Leitlinien für die Lagerung, den Verkauf, die Auslage und die Verschreibung von Medikamenten bereitzustellen.
-
1988 wurde das Gesetz reformiert, um die GMP-Prinzipien der WHO zu integrieren.
-
2005 wurde “Schedule Y” des Gesetzes (die Leitlinien für klinische Studien enthaltende Kategorie) aktualisiert, um präzisere Standards und Anweisungen für die Durchführung klinischer Studien in Indien aufzunehmen.
-
The Patents Act (1970): Ermöglichte die Entwicklung der indischen Generika-Industrie, indem nur Verfahrenspatente, nicht Produktpatente anerkannt wurden.
-
Modernisierung und globale Expansion (2000er bis heute):
Wurde ein bedeutender Akteur im Bereich “Contract Research and Manufacturing Services” (CRAMS)
-
Bildung von Regulierungsbehörden wie der Central Drugs Standard Control Organization (CDSCO) und der National Pharmaceutical Pricing Authority (NPPA) zur Verbesserung der Arzneimittelqualität und Sicherheitsstandards und zur Sicherstellung der Erschwinglichkeit
-
Änderungen in Indiens Patentrecht aufgrund der Einführung des TRIPS-Abkommens (Trade-Related Aspects of Intellectual Property Rights) der Welthandelsorganisation, die den Ansatz zur Generika-Produktion beeinflussten.
-
The New Drugs and Clinical Trials Rules (2019): Reformierten den regulatorischen Rahmen durch Reduzierung der Komplexität in der klinischen Forschung und Verbesserung der Prozesse für die Arzneimittelregistrierung und Post-Marketing-Überwachung.
Russland
-
Sowjetische Regulierung (Nach der Revolution von 1917 — 1930): Nach der Revolution von 1917 errichtete die Regierung ein staatliches Monopol, das alle Aspekte der pharmazeutischen Produktion, Distribution und Qualitätssicherung kontrollierte, einschließlich der Gründung der Pharmacopoeia Commission 1923 (die die pharmazeutische Qualität kontrollierte) und der umfassenden Verwaltung pharmazeutischer Aktivitäten unter dem Volkskommissariat für Gesundheit bis 1930.
-
Neuere Regulierung (2010-11): Erlass von Bundesgesetzen zur Verbesserung der Gesundheitsversorgung und Arzneimittelversorgung, zusammen mit dem Gesetz “Über den Verkehr von Arzneimitteln” im Jahr 2010, um die Qualität und Sicherheit von Medikamenten zu gewährleisten, was die sich entwickelnde Gesundheitslandschaft widerspiegelt.
Simbabwe
-
The Drugs and Allied Substances Control Act (1969): Ein Gesetz, das die systematische Kontrolle und Verwaltung von Arzneimitteln im Land begann.
-
Gründung der Medicines Control Authority of Zimbabwe oder MCAZ (1997): Geschaffen zur Aufsicht und Regulierung von Arzneimitteln im Land.
-
Vorschriften für homöopathische Mittel (2015): Ein Gesetz, das eine offizielle Aufhebung oder Widerrufung einer früheren Reihe von Vorschriften für homöopathische Mittel in Simbabwe darstellt.
-
Übernahme der Good Distribution Practice (GDP)-Leitlinien (2018): Kürzlich übernahm die MCAZ GDP-Leitlinien, um die Qualität und Sicherheit medizinischer Produkte in allen Aspekten des Vertriebsprozesses sicherzustellen.
Was ist mit Ländern, die kein unabhängiges Arzneimittelregulierungssystem haben?
Trotz dieser Strategien hat die Forschung gezeigt, wie Entwicklungsländer immer noch auf Widerstand bei der Übernahme globaler Standards für pharmazeutische Qualität und regulatorische Infrastruktur stoßen. Länder, die kein eigenes unabhängiges Regulierungssystem für Arzneimittel haben (in der Regel kleinere Länder ohne eigene explizite Regulierungsbehörden), nutzen bestimmte Strategien:
-
WHO-Präqualifizierung: Diese Länder verlassen sich regelmäßig auf das WHO-Präqualifizierungssystem, das die Qualität, Sicherheit und Wirksamkeit wichtiger Medikamente analysiert und sicherstellt.
-
Das Kooperationsverfahren zur beschleunigten Registrierung: Unter Federführung der WHO beschleunigt es die Produktregistrierung in zahlreichen Ländern, indem es die Arbeit strenger Regulierungsbehörden nutzt.
-
Bezugnahme auf benachbarte Rahmenbedingungen: Einige Länder übernehmen oder beziehen sich auf die regulatorischen Rahmenbedingungen benachbarter Länder oder Regionen mit etablierteren Systemen.
-
Verlass auf internationale Leitlinien: Länder können internationale Leitlinien von Organisationen wie der ICH und der WHO verwenden.