
Vergleich von KI-Laboren und Pharmaunternehmen
Im letzten Jahr habe ich viel darüber nachgedacht, was der Bereich der KI von der Pharmaindustrie lernen könnte. Als Arzt, der in klinischen Studien gearbeitet hat, bin ich mit den dortigen Prozessen bestens vertraut und habe gelernt, sie im Laufe der Zeit tief zu schätzen. Ich habe auch einige Monate damit verbracht, mich in KI weiterzubilden - vom Verständnis der Funktionsweise von Transformern und dem Besuch von Konferenzen bis hin zum Projektmanagement von ML-Läufen und Cybersecurity-Workshops.
Hier stelle ich KI-Labore und Pharma gegenüber, zeige einige meiner bevorzugten Beispiele für Diskrepanzen und diskutiere rigorosere Bewertungen von KI-Modellen, inspiriert von klinischen Studienrahmenwerken.
Danke an LightSpeed Grants für die Bereitstellung von Fördermitteln zur Untersuchung dieses Themas und an die vielen Personen, mit denen ich diese Gedanken im letzten Jahr diskutiert habe.
KI-Labore vs. Pharma
“Die Branche produziert Produkte, die großen Nutzen bringen und schwere Risiken für Einzelpersonen darstellen können. Umfangreiche technische Expertise ist für die Produktentwicklung erforderlich, und es fallen hohe Kosten ($100M+) an.”
Diese Aussage trifft sowohl auf KI-Labore als auch auf Pharmaunternehmen zu. Darüber hinaus gravitieren Regulierungsbehörden zunehmend zu einer ähnlichen Dichotomie von “weitgehend unreguliert” und “streng reguliert” im KI-Bereich, ähnlich wie es bereits bei der Arzneimittelregulierung der Fall ist. Die Verwendung von Indikatoren wie Anwendungskontext und investierten Rechenressourcen (~10^26 FLOPs) ähnelt der Unterscheidung zwischen der Regulierung von Arzneimitteln und Nahrungsergänzungsmitteln.
In den folgenden Tabellen stelle ich F&E-Prozesse und Stakeholder gegenüber. Frühe Forschungs- und Testphasen sehen recht ähnlich aus. Später im F&E-Prozess werden deutliche Unterschiede sichtbar, wie der häufig diskutierte Unterschied im Weg zur Zulassung/Bereitstellung/Lizenzierung, der für KI (zu diesem Zeitpunkt) weit weniger systematisiert ist und oft komplette Arbeitsbereiche vermissen lässt.
Es gibt viele weitere Unterschiede. Ein Kernunterschied besteht darin, dass die Phasen der Arzneimittelentwicklung darauf ausgelegt sind, die Sicherheit von Wirkstoffkandidaten rigoros zu testen. Sicherheit (Safety) wird definiert als die Verhinderung, dass ein System oder Produkt seine Umgebung auf unerwünschte oder schädliche Weise beeinflusst, typischerweise zum Schutz von Menschenleben, der natürlichen Umwelt oder von Vermögenswerten. Der Arzneimittelentwicklungsprozess ist nicht darauf ausgelegt, Bedrohungsmodelle zu identifizieren und zu bekämpfen, bei denen jemand ein Medikament absichtlich gegen andere einsetzen würde, d.h. Sicherheitsrisiken (Security). Security zielt darauf ab, oft feindlich gesinnte Akteure oder Bedingungen daran zu hindern, ein System auf unerwünschte oder schädliche Weise zu beeinflussen, z.B. Produktwaffnung, autonome Replikation, Sicherheitsverletzungen oder breite gesellschaftliche Risiken. Tatsächlich kam ein Bericht des National Research Council von 2010 zu dem Schluss, dass die Vorhersage von Eigenschaften wie Virulenz oder Pathogenität - mit dem für regulatorische Zwecke notwendigen Grad an Sicherheit - in absehbarer Zukunft unmöglich sein würde, um besser mit den Risiken von biotechnologisch hergestellten potenziellen Pandemieerregern umzugehen.
Während die Sicherheit von KI-Systemen eindeutig ein Anliegen ist, sind die meisten Experten noch besorgter über deren Security-Implikationen, d.h. wie dieses Werkzeug missbraucht werden kann. Die KI-Branche kann umfassend von der Arzneimittelentwicklung über die Erkennung und das Management unerwünschter Ereignisse lernen, aber wenn es darum geht, Gruppen oder sogar ganze Gesellschaften vor Technologiemissbrauch zu schützen, muss die KI-Branche anderswo nach Orientierung suchen.
Wenn überhaupt, bedeutet das, dass KI über das hinausgehen sollte, was in der Arzneimittelentwicklung erwartet wird, oder?

Trotz deutlicher Unterschiede haben viele Autoren über verschiedene Lehren geschrieben, die insbesondere aus der FDA für die Regulierung und Lizenzierung in der KI-Branche gezogen werden könnten.
Die vier Lehren, die eng mit der Pharmabranche (im Gegensatz zu Arzneimittelregulatoren) verbunden sind und selten diskutiert werden, sind:
-
Budget
-
Wissenschaftlicher Ansatz
-
Geltungsbereich
-
Kontrollmechanismen im Ökosystem
Zentrale Verbesserungsbereiche für KI
| Bereich | Arzneimittelentwicklung | KI |
|---|---|---|
| (Kontext) | Die Arzneimittelentwicklung und andere Branchen haben den größten Teil eines Jahrhunderts damit verbracht, Methoden zur Bewertung, Prüfung und Qualitäts- und Sicherheitsgewährleistung ihrer Produkte zu entwickeln. Best Practices der Branche haben über die Jahre enorme Fortschritte gemacht. Allerdings wurden diese Standards nicht an einem Tag aufgebaut, und das Fehlen angemessener Maßnahmen hatte schwerwiegende Folgen für Unternehmen und Patienten. | KI ist ein junges Feld mit wenig bis keiner Erfolgsbilanz bei sicherheitsbezogenen Maßnahmen. Angesichts der begrenzten Übernahme aus verwandten Bereichen (und minimaler Aufmerksamkeit aus der Wissenschaft) sollten wir sehr besorgt sein, wenn KI-Labore die gesellschaftlichen Erwartungen nicht erfüllen, die wir an andere Hochrisikosektoren haben. |
| Budget | Heute werden etwa 50-90% der gesamten F&E-Kosten in der Arzneimittelentwicklung in Qualitäts- und Sicherheitsmaßnahmen investiert (einschließlich Gemeinkosten für Good Manufacturing Practices, Tierversuche, klinische Studien usw.). | KI-Unternehmen scheinen nur niedrige einstellige Prozentsätze für Assurance-Maßnahmen auszugeben (und relativ gesehen immer weniger aufgrund steigender Trainingskosten). Dies stellt eine Umkehrung dessen dar, was wir in reifen Industrien wie Pharma oder Luftfahrt sehen. |
| Kontrollmechanismen im Ökosystem | Die Arzneimittelentwicklung findet innerhalb eines Ökosystems statt und wird bei weitem nicht von einer einzigen Entität durchgeführt. Dies ermöglicht zahlreiche gegenseitige Kontrollen. | KI-Unternehmen sind größtenteils autonom und haben wenige Kontrollmechanismen. |
| Geltungsbereich | Tests hängen vom Geltungsbereich ab und müssen wiederholt werden, wenn sich der Bereich oder das Produkt ändert. Man muss sehr vorsichtig sein, wenn man ein Medikament außerhalb der Verteilung des Getesteten anwendet. | Die Anwendung von KI scheint nicht anzuerkennen, wenn sie “außerhalb des Geltungsbereichs” liegt. Sicherheitsergebnisse werden oft übermäßig verallgemeinert. |
| Wissenschaftlicher Ansatz | Arzneimittelbewertungen basieren auf der Quantifizierung akzeptabler Risiken und der Gewinnung statistisch zuverlässiger Informationen über reale Risiken. | KI-Labore verfolgen derzeit einen eher qualitativen Ansatz zur Risikobewertung ohne angemessene Hypothesentests. |
Budget: Geld dort investieren, wo Qualität und Sicherheit sind
Dies war eine der überzeugendsten Erkenntnisse für mich. Kurz zusammengefasst: 50-90% der gesamten Arzneimittelentwicklungskosten fließen in Sicherheitstests und Qualitätssicherung. Rigoroses Testen, einschließlich des Aufgebens unsicherer oder unwirksamer Produkte, macht die Arzneimittelentwicklung so teuer - nicht die ingenieurtechnischen Herausforderungen bei der Herstellung des Medikaments selbst. Mit Investitionen von Hunderten von Millionen in Sicherheitsbewertungen stellen diese wahrscheinlich den größten einzelnen F&E-Kostenfaktor in einem Arzneimittelentwicklungsprogramm dar, gefolgt von Wirksamkeitsbewertung, gescheiterten Kandidaten und Prozessentwicklung/Herstellung.
Um einige Zahlen zu nennen: F&E-Ausgaben pro zugelassenem Medikament variieren zwischen unter 1 Milliarde und über 2 Milliarden Dollar. Gescheiterte Kandidaten machen ein Drittel dieser Kosten aus. Prozessentwicklung und Herstellung verbrauchen etwa 15% des F&E-Budgets von präklinischen Studien bis zur Zulassung. Der Großteil (~60%) der Ausgaben für erfolgreiche Kandidaten entsteht während der klinischen Tests, wobei die Kombination aus Phase-1- und Phase-4-Studien am kostenintensivsten ist. Offen gesagt ist es schwierig, die Kosten zu disambiguieren, da praktisch alles um das zentrale Dogma der Herstellung eines sicheren und zuverlässigen Medikaments kreist.
Für eine Arzneimittelzulassung über 1 Milliarde Dollar:
-
$330M (33%): Gescheiterte Kandidaten, die hauptsächlich aus Gründen der Wirksamkeit oder Sicherheit aufgegeben wurden
-
$100M (10%): Prozessentwicklungs- und Herstellungskosten
$66M (6%): Strenge Qualitäts- und Testkriterien in GMP (2/3) und tatsächliche Herstellungskosten (1/3) (Schätzung aus Expertenerfahrung)
-
Erste GMP-Charge im kleinen Maßstab (bis Phase 1): $3-10M GMP-Charge im mittleren/großen Maßstab (Phase 2 bis Zulassung): $25-50M
-
$225M (22,5%): Präklinische Entwicklungsarbeiten, einschließlich vieler Iterationen von
In-vitro bei weniger als 0,5M,
-
Tierversuchen (0,5-5M) und
-
ersten Non-GMP-Arzneimittelchargen im kleinen Maßstab bei 0,5-1,5M
-
$335M (33,5%): Klinische Studien
Die Kosten variieren dramatisch, können aber mit 4, 13, 20 und 20 Millionen Dollar angegeben werden.
-
Durchschnittliche Anzahl der Studien: Phase 1 (1,7), Phase 2 (2,0), Phase 3 (2,8), Phase 4 (3,2)
-
Zusammengerechnet ergeben sich durchschnittliche Kosten von $155M
-
Kapitalopportunitätskosten machen aufgrund der jahrzehntelangen Entwicklungszeitlinie 25-50% der Kosten in jeder Kategorie aus:
Präklinische Phase: ~31 Monate
- Klinische Phase: 5,9-7,2 Jahre (nicht-onkologisch), 13,1 Jahre (Onkologie) Individuelle Studiendauern: Phase 1 (1,6 Jahre), Phase 2 (2,9 Jahre), Phase 3 (3,8 Jahre)
Betrachtet man die Arzneimittelentwicklungskosten, so machen Sicherheit und Qualitätssicherung über 50% der gesamten F&E-Ausgaben aus. Bei der Analyse klinischer Studien, Tierstudien in präklinischen Phasen, gescheiterter Kandidaten und GMP-Gemeinkosten reicht das Verhältnis von Entwicklungs- zu Sicherheitskosten von 1:1 bis 1:10, je nach Therapiebereich und verwendeten Definitionen. Neuartige Arzneimitteldesigns erfordern zusätzliche obligatorische Sicherheitsmaßnahmen aufgrund ihres unerforschten Charakters, was bedeutet, dass Sicherheit und Qualitätssicherung 50-90% der Gesamtkosten ausmachen können.
In der Luftfahrt zeigt meine kurze Untersuchung der F&E-Kostenaufschlüsselung für Passagierflugzeuge Gesamtkosten von etwa dem Zehnfachen der Arzneimittelentwicklung, rund $10+ Milliarden pro neuem Flugzeug. Sicherheit und Qualitätssicherung sind in jeden F&E-Schritt integriert, und der Test-/Zertifizierungsprozess allein erstreckt sich über mehrere Jahre und verschlingt Hunderte von Millionen Dollar. Das Verhältnis erscheint näher an 1:1 zwischen Test/Qualitätssicherung und tatsächlichen Herstellungskosten, obwohl dies einer tieferen Untersuchung bedarf. (Eine hilfreiche Intuition: Ein neues Passagierflugzeug kostet im niedrigen $100M-Bereich, also 1-5% der Entwicklungskosten)
KI-Entwickler scheinen am entgegengesetzten Extrem zu stehen, mit geschätzten 95%+ der gesamten Bereitstellungskosten für das, was im Wesentlichen Herstellung ist - das Training des Modells. Zum Beispiel enthielten die geschätzten $100M Kosten von ChatGPT nur einen trivialen Betrag für Sicherheitsmaßnahmen. Brancheninsider bestätigen, dass Sicherheitstests typischerweise nur niedrige einstellige Prozentsätze an Investitionen erhalten.
Angesichts der erhöhten Einsätze bei KI erscheint ein Mindestverhältnis von 1:10 (Trainingskosten zu Assurance-Maßnahmen) für KI-Modelle angemessen - und möglicherweise mehr. Allerdings bleibt selbst dieses vorgeschlagene Verhältnis hinter den Empfehlungen besorgter KI-Experten zurück, die mindestens ein Drittel der F&E-Budgets für Sicherheit und ethische Nutzung fordern. Es liegt auch unter den proklamierten Sicherheitsbudgets einiger KI-Labore, wie dem ehemaligen Superalignment-Team von OpenAI mit 20%.
Ökosystem der gegenseitigen Kontrolle: Mehrparteien-Entwicklung mit Verantwortlichkeit der Stakeholder
Die Arzneimittelentwicklung verfügt über viele Mechanismen, um unabhängige Parteien in den Prozess einzubinden und ihnen Einfluss zu geben. Viele Produktions- und Herstellungsdienstleistungen werden von externen Anbietern erbracht, Auftragsforschungsinstitute überwachen Tests, Apotheker handhaben das Produkt und Ärzte rekrutieren (und behandeln später) Patienten. Jeder Einzelne von ihnen ist für seine Arbeit verantwortlich und muss sicherstellen, dass Best Practices eingehalten werden. Andernfalls sind sie rechtlich haftbar, können ihre Betriebslizenz verlieren und sogar im Gefängnis landen.
In der folgenden Tabelle habe ich versucht, verschiedene Kategorien von Stakeholdern zwischen Arzneimittelentwicklung und KI gegenüberzustellen. Was die Erledigung von Aufgaben betrifft, ist die Synchronisation zwischen mehr Personen sicherlich eine Herausforderung. Aber die Aufteilung der Verantwortung auf verschiedene Akteure und deren gesetzliche Rechenschaftspflicht erscheint als ein wesentlich widerstandsfähigerer Weg, ein Feld voranzubringen. Anreize können besser gegen Rücksichtslosigkeit und Betrug ausgerichtet werden.
Eine Welt, in der KI-Labore nicht nur ein Rechenzentrum mit genügend Strom und GPUs finden müssen, sondern sich auch intensiv mit mehreren Ausschüssen, unabhängigen Gremien, Spezialisten für sensible Anwendungen usw. auseinandersetzen müssen, wäre für mich weitaus attraktiver.
Tabelle: Stakeholder in der Arzneimittelentwicklung vs. KI-Entwicklung mit hohem Risiko
Im Folgenden werden die “Akteure” in der Arzneimittelentwicklung mit denen in der Entwicklung von KI-Modellen mit hohem Risiko verglichen. Zu beachten ist, dass für jeden Akteur der Arzneimittelentwicklung plausible Äquivalente in der KI-Entwicklung mit hohem Risiko existieren, die meisten jedoch noch nicht etabliert sind, obwohl erste Bemühungen im Gange sind. Die Einrichtung unabhängiger Gremien, Zertifizierungen und öffentlicher Berichtssysteme wurde zuvor als entscheidend für die KI-Governance identifiziert.
| Bereich | Kategorie | Umsetzung in der Arzneimittelentwicklung | Umsetzung in der Entwicklung von KI-Modellen mit hohem Risiko |
|---|---|---|---|
| Regulierungsbehörde | Internationale Richtlinienentwicklung | ICH / OECD / WHO Good X Practice (GxP, x = variabel) Richtlinien mit lokaler Umsetzung, z.B. Good Clinical Practice (GCP) - für Design und Durchführung klinischer Studien; Good Laboratory Practice (GLP) - für Design und Durchführung nichtklinischer (Tier-)Studien; Good Manufacturing Practice (GMP) - für die Herstellung des Arzneimittels. “GMP-Material” bedeutet ein unter Einhaltung der GMP-Standards hergestelltes Arzneimittel. | z.B. NIST AI Risk Management Framework und ISO/IEC FDIS 23894. |
| Lokales Recht und regulatorische Leitlinien | Die Einhaltung dieser Standards ist durch FDA-Vorschriften für US-Einreichungen vorgeschrieben (z.B. 21CFR210 für GMP, 21CFR58 für GLP, mehrere Teile von 21CFR für GCP) und oft auch von anderen Ländern. | EU AI Act, Leitlinien für ethische KI und ähnliche Gesetze | |
| Regulierungsbehörden | FDA, EMA, … | Unklar | |
| Unabhängige Gremien | Unabhängige Beratungsausschüsse | Z.B. Expertengruppen, die Bewertungen für Regulierungsbehörden erstellen | Kein Standard |
| Unabhängige Ethikkommissionen | Institutional Review Boards (IRBs) | Kein Standard | |
| Unabhängige Sicherheitsausschüsse | Sicherheitsdatenbanken und Data and Safety Monitoring Boards (DSMBs) | Kein Standard | |
| Unabhängige Prüfer | Überprüfung durch unabhängige externe Fachleute sowie Bundesbehörden | Kein Standard | |
| Öffentliche Beteiligung | Patientenorganisationen in jeweiligen Gremien | Kein Standard | |
| Non-Profits | Öffentliche Interessenvertretungen und gemeinnützige Überwachungsgruppen | GovAI und Think Tanks wie RAND | |
| Berichtswesen | Öffentliche Datenbanken | Clinicaltrials.gov, andere nationale Studienregistrierungen | Kein Standard |
| Veröffentlichung von Ergebnissen in Fachzeitschriften | Medizinische Fachzeitschriften (obligatorische Publikationsstrategie) | Kein Standard, Arxiv (freiwillig) | |
| Unternehmen | Sponsor | Pharmaunternehmen (oft der Sponsor) | KI-Labor |
| Dienstleister | Operative Dienstleistungen | Auftragsforschungsinstitute (CROs) für Projektmanagement | Kein Standard |
| Fachexperten | Berater für Krankheiten, Regulierung, Anwendungen usw. | Z.B. Berater für chemische/biologische Dual-Use-Themen und andere schädliche Anwendungen | |
| Herstellung | Contract Development and Manufacturing Organization (CDMO) | Rechenzentren / Cloud-Anbieter / Energieversorger | |
| Frühe Produktbewertung | Tierversuchsanbieter, spezialisierte Labore | Organisationen wie METR | |
| Schulungs- und Zertifizierungsorganisationen | Medizinische/pharmazeutische Lizenzen, GxP-Schulung, Audits usw. | Kein Standard | |
| Logistik-/Vertriebsanbieter | Lieferkette/Apotheken | Cloud-Anbieter, die Modelle betreiben (z.B. legaler Zugang zu KI-Modellen nur über zertifizierte Anbieter) | |
| Erweiterte Produktbewertung | Studienzentren | Kein Standard. Potenziell: Unternehmen mit speziell zertifizierten Cybersecurity-Systemen für kontrollierte Real-World-Evaluierungen | |
| Lokal verantwortlich | Arzt als Hauptprüfer (PI) | Kein Standard. Potenziell: KI-Beauftragte in Unternehmen | |
| Studienteilnehmer | Patienten | Kunden / IT-Systeme / Datenstrukturen / usw. | |
| Praxisanwendung | Realtests | Phase-4-Studien | Öffentliche Beta von ChatGPT und anderen LLMs |
| Überwachung in der Praxis | Z.B. FDAs Adverse Event Reporting System (FAERS) | Kein Standard; OECD-Vorschlag für Incident-Reporting-Datenbank | |
| Kunden | Patienten | Kunden | |
| Unbeteiligte Dritte | Zugangskontrolle über Ärzte. Für die Arzneimittelentwicklung gibt es kaum bis keine Geschichte von Schäden an Nicht-Kunden | Starker Verdacht auf potenzielle Hochrisiko-Komplikationen für unbeteiligte Dritte |
Geltungsbereich: Implikationen verschiedener Kunden und laufender Modifikation
Zugelassene Medikamente haben immer eine vorgesehene Zielpopulation. Die Verschreibung außerhalb dieser Population betritt eine rechtliche Grauzone und wird mit äußerster Vorsicht angegangen. Ebenso erfordern Medikamentenmodifikationen (und sogar Generika, die den Wirkstoff replizieren) umfassende neue Studien, um zu beweisen, dass a) vergangene Evidenz relevant bleibt und b) neue Evidenz die Sicherheit in einer ähnlichen Zielpopulation demonstriert. Dies erzeugt eine inhärente Spannung zwischen der Bereitstellung der neuesten Innovationen für alle potenziellen Patienten weltweit und der Aufrechterhaltung von Sicherheitsstandards für jede Zielgruppe und Version.
KI-Entwickler scheinen ihre Sicherheitsbewertungen unzureichend einzugrenzen und an Kunden und in Kontexte zu liefern, die weit von ihren Testszenarien entfernt sind, ohne angemessene Verfolgung der Konsequenzen.
Während die Sicherheitstests generativer KI-Modelle aufgrund ihrer breiten Fähigkeiten als ungewöhnlich komplex gelten, sollte dies nicht dazu führen, Sicherheitstests aufzugeben. Stattdessen deutet es darauf hin, dass die Fähigkeiten zu breit sind und eingegrenzt werden müssen, um eine schlüssige Sicherheitsdatenerhebung zu ermöglichen. Die Lösung besteht darin, Anwendungsfälle für generative KI-Modelle einzuschränken. Viele Wirkstoffkandidaten zeigen ebenfalls weitreichende potenzielle Vorteile bei verschiedenen Krankheiten oder sogar bei gesunden Personen. Aufgrund der Kosten für den Aufbau von Produktsicherheitsvertrauen können Unternehmen sie jedoch nur für gezielte Populationen vermarkten, in denen überzeugende Evidenz für Sicherheit (und Wirksamkeit) gesammelt wurde.
Ob für die Gesellschaft insgesamt, Unternehmen oder bestimmte Einzelpersonen - verschiedene Anwendungsfälle erfordern verschiedene Arten und Mengen schlüssiger und zuverlässiger Evidenz zur Bestimmung der Produktsicherheit, entweder durch Unternehmenstests oder regulatorische Aufsicht. Dies erfordert einen strukturierten Ansatz und eine externe Validierung von Versuchsplanung, -durchführung und -analyse.
Wissenschaftliches Testen: KI-Sicherheitsstudien zur zuverlässigen Bewertung gefährlicher Fähigkeiten - Skalierung zählt
In der Arzneimittelentwicklung teilen alle Stakeholder eine zentrale Obsession: Menschen keinen Schaden zufügen. Dies gilt sowohl für Studienteilnehmer als auch für die Millionen, die das Medikament schließlich erhalten werden. Massive Ressourcen werden investiert, um die Sicherheit zu gewährleisten, wobei jeder Aspekt der Entwicklung, des Testens und der Einführung so gestaltet ist, dass vordefinierte Bedingungen erfüllt werden.
Es gibt verschiedene Möglichkeiten, akzeptable Risiken zu quantifizieren, und die Risikotoleranz hängt immer vom Kontext und den potenziell positiven Auswirkungen ab. Zum Beispiel unterscheidet sich das akzeptable Risikoprofil für ein potenziell lebensrettendes Medikament bei einem todkranken Patienten erheblich von dem eines Impfstoffs gegen ein mildes Virus bei gesunden Personen. Die schwerwiegendsten unerwünschten Ereignisse in der Arzneimittelentwicklung werden als “schwerwiegende unerwünschte Ereignisse” (SAE) bezeichnet und umfassen sieben spezifische Ereignistypen (siehe hier für die Definition). Für die meisten Medikamente können bereits einstellige Vorkommen dieser Ereignisse ein gesamtes Entwicklungsprogramm stoppen oder das Medikament für bestimmte Patientenpopulationen einschränken. Ereignisraten schwerwiegender unerwünschter Ereignisse zwischen 1:100 und 1:10.000 gelten oft als inakzeptabel.
Diese Strenge besteht, weil die Einführung von Medikamenten in einer gesamten Population potenziell Tausende von Todesfällen oder anderen SAEs verursachen könnte - Ergebnisse, die in den meisten Fällen als absolut inakzeptabel gelten. Die Verhinderung solcher Situationen erfordert rigorose Evidenzerhebung, Überwachung und Qualitätssicherung.
Die Obergrenze des in der Arzneimittelentwicklung vorgesehenen Schadens scheint die Untergrenze dessen zu sein, was KI-Experten als katastrophale Risiken betrachten. Über unmittelbare individuelle Schäden hinaus erkennen KI-Experten potenzielle katastrophale gesellschaftliche Risiken an, die von “Tausenden von Todesfällen oder Hunderten von Milliarden Dollar an Schäden” (Anthropics RSP) bis hin zu Auslöschungsereignissen reichen.
Aktuelle primäre Bedrohungsmodelle konzentrieren sich auf:
-
Missbrauch durch böswillige Akteure, die KI nutzen, um Schaden durch Cyber-, Bio-, Nuklear- oder soziale Unruhen zu verursachen
-
Ähnliche Bedrohungen durch autonome, unkontrollierbare KI-Agenten
Aktuelle Rahmenwerke zur Bewertung von Risiken von Frontier-KI-Modellen umfassen typischerweise Teams von 3-10 Fachexperten, die versuchen, bestimmte Fähigkeiten zu elizitieren, verglichen mit fairen Vergleichsmaßstäben wie Suchmaschinen oder menschlicher Leistung (wie in biologischen Evaluierungsberichten und verschiedenen System Cards zu sehen).
Während einzelne Teams verschiedene Prompts und Pfade testen, garantiert die Unfähigkeit eines einzelnen Teams, gefährliche Fähigkeiten mit gegebenen Ressourcen zu elizitieren, nicht, dass andere gleich fähige Teams nicht erfolgreich wären - wenn auch nur durch Zufall. Die Elizitierung von Fähigkeiten in KI-Modellen ist ein junges Feld, und laufende Demonstrationen von Jailbreaks und anderen Angriffen durch Amateure unterstreichen die Bedeutung von Skalierung beim Testen.
Wenn wir behaupten wollen “Personen vom Typ X können diese Fähigkeit bei gegebenen Y Ressourcen nicht elizitieren”, brauchen wir Experimente mit akzeptablen Ereignisraten (z.B. 1:1000) und Powergrenzwerten (z.B. 90% Power), die mehrfach unabhängig wiederholt werden.
Mit ausreichender Wiederholung (geleitet durch Fallzahlberechnungen) könnte die Nichtentdeckung eines Teams, das eine gefährliche Fähigkeit elizitiert, Aussagen wie diese stützen: “Wir sind zu 95% sicher, dass weniger als 1:1000 Teams von X Personen mit Ressourcen Y in der Lage sein werden, diese Fähigkeiten aus dem Modell zu elizitieren.” Obwohl dies nicht vollständig vor katastrophalen Risiken schützt, liefert es wesentlich zuverlässigere Daten als Bewertungen von Mitarbeitern des KI-Labors, die offensichtliche Interessenkonflikte haben.
KI-Labore sollten Mitigationsmaßnahmen für Frontier-Modelle bewerten und implementieren, um statistisch zuverlässige Daten darüber zu erhalten, ob Einzelpersonen oder Teams auf verschiedenen Expertise- und Ressourcenniveaus ihre Modelle missbrauchen können. Ähnliche Evidenz wird für gefährliche Autonomierisiken benötigt, obwohl sich diese Diskussion auf Missbrauchskontexte konzentriert. Zum Verständnis verschiedener Bedrohungsakteure definiert RANDs jüngster Bericht zur Sicherung von KI-Modellgewichten fünf operative Kapazitätsstufen (OC) von OC1: Amateurniveau (~$1.000) bis OC5: Staatliches Akteursniveau ($1 Milliarden Budget). Diese Ressourcenniveaus sind auch für Bewertungen gefährlicher Fähigkeiten relevant.
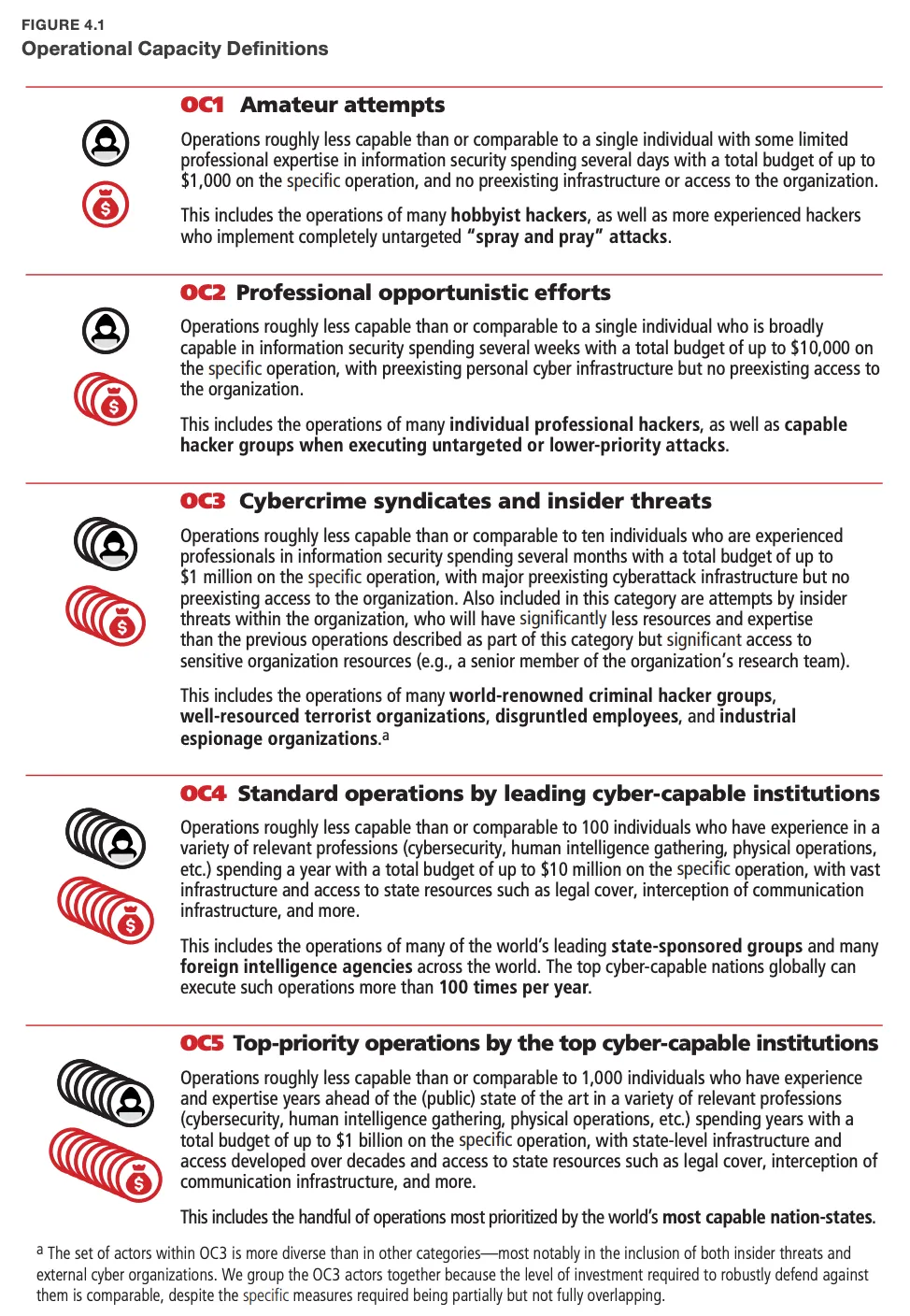
Um zuverlässige Hypothesentests in Missbrauchsszenarien zu demonstrieren, betrachten wir die globale Verteilung von Akteuren über operative Kapazitätsstufen:
-
OC1 (Amateur): 100.000 Akteure
-
OC2: 10.000 Akteure
-
OC3: 1.000 Akteure
-
OC4: 100 Akteure
-
OC5 (Staatlich): 2 Akteure
Unter Verwendung der Dreierregel in der Statistik benötigt man, um unerwünschte Fähigkeitserlangung auf jeder OC-Stufe zuverlässig auszuschließen, ungefähr die dreifache Anzahl von Testinstanzen. Zum Beispiel würden 300.000 OC1-Versuche es ermöglichen, mit 95% Konfidenz zu sagen, dass weniger als 1:100.000 Amateur-Codierer mit maximal $1k Ressourcen gefährliche Fähigkeiten elizitieren können.
Die Elizitierung gefährlicher Fähigkeiten könnte durch aggregierte Scores über Domänen oder einzelne Metriken gemessen werden, mit sorgfältiger Berücksichtigung geeigneter primärer Endpunkte.
Wichtig ist, dass diese Teststärke erreichbar ist: Lakera hat dies demonstriert, indem sie “Gandalf” entwickelten, ein Online-Jailbreaking-Spiel, das Benutzer herausfordert, immer schwierigere clevere Prompts zu entwickeln. Ihre Community hat insgesamt 25 Jahre in über 1 Million Sitzungen investiert, um ihr System zu jailbreaken.
In ihrer Responsible Scaling Policy definiert Anthropic, dass sie Akteure, die in der Lage sind, gefährliche Fähigkeiten mit “1% der gesamten Trainingskosten” zu elizitieren, als besorgniserregend betrachten würden, sodass sie substanzielle zusätzliche Sicherheitsmaßnahmen einleiten würden. 1% der gesamten Trainingskosten entspricht ungefähr OC3, benchmarkt bei ~1 Million Dollar. Wenn wir mit 95% Konfidenz sicherstellen wollten, dass weniger als 1:1.000 dieser Gruppen dazu in der Lage sind, bräuchten wir ~3.000 Versuche, sehr ähnlich der Mindestanzahl von Teilnehmern in der Impfstoff-Arzneimittelentwicklung, wo Daten zum Nachweis unerwünschter Ereignisse von 1:1.000 vorgelegt werden müssen. Dies wäre gleichbedeutend mit der Durchführung einer Phase-3-Studie, aber wahrscheinlich mit 100-fachen Kosten (ungefähr $30M/Studie vs. $3B). Wichtig ist, man könnte argumentieren, dass, obwohl 1.000 solcher Akteure weltweit existieren, nicht alle den Missbrauch des Modells verfolgen / so viel Geld dafür ausgeben werden. Man könnte annehmen, dass das skizzierte Experiment von 3.000 Versuchen mit durchschnittlich $1M pro Versuch auch OC4-Akteure (Akteure mit $10M Finanzierung) einschließen würde. Ähnlich wie in der Arzneimittelentwicklung würde man davon ausgehen, dass mehrere solcher Studien notwendig sein könnten, insbesondere für allgemeine KI-Modelle. Allgemeine Modelle haben viele Bedrohungsmodelle, was das Testen aller äußerst schwierig macht. Die Einschränkung der Modellfähigkeiten, z.B. durch reines Training auf Code oder rein auf Biologie usw., würde die Notwendigkeit mehrerer Testpfade beseitigen.
Missbrauch durch OC5-Akteure liegt wahrscheinlich außerhalb des Umfangs von Sicherheitstests für Modelle, da die Finanzierung (>= $1B) dieser Akteure ausreicht, um selbst Frontier-Modelle zu erstellen. Darüber hinaus würden Studien auf OC3- und OC4-Niveau Indikatoren für entscheidende Maßnahmen liefern, die auch für noch besser ausgestattete Akteure gelten würden. Zusätzlich könnten Sicherheitspuffer als Proxy dafür dienen, was für staatliche Akteure als möglich angenommen wird (z.B. Verschiebung des Schwellenwerts zum konservativeren Ende).
Die oben genannten Vorschläge erfordern eine erhebliche Investition in Testinfrastruktur. Ähnlich wie bei den Interaktionen zwischen Pharmaunternehmen, Auftragsforschungsinstituten und qualitätsgesicherten Studienzentren, die Patienten rekrutieren, müssten Frontier-KI-Labore wahrscheinlich mit Unternehmen zusammenarbeiten, die eine Plattform bereitstellen, um sicher und mit sehr klaren Überwachungs- und Berichtsanforderungen mit qualitätskontrollierten Teams und Partnern zusammenzuarbeiten.
Anhang
Vergleich des Entwicklungsprozesses von Arzneimitteln und KI-Modellen
Diese Tabelle listet alle wesentlichen Schritte des Arzneimittelentwicklungsprozesses auf und ordnet entsprechende Schritte und Terminologie aus dem KI-Modellentwicklungsbereich zu. In den frühen Phasen gibt es erhebliche Überschneidungen, während in späteren Phasen kein Äquivalent für klinische Tests bei KI-Modellen existiert.
| Arzneimittelentwicklung | Meilensteine | KI-Modellentwicklung |
|---|---|---|
| Literaturrecherche zur Zielpopulation und Krankheit; Patientenprobenahme; Expertenberatung | Discovery | Ziel: Allgemeine künstliche Intelligenz |
| In-silico-/In-vitro-Prototyping. Identifikation des Produkts. Korrespondenz mit Regulierungsbehörden über die beste Bewertung und Bestimmung von Sicherheit/Wirksamkeit | Forschung | Prototyping (Testläufe, Hypothesentests, frühe Algorithmusbewertungen) |
| Herstellung erster Wirkstoffsubstanz durch Prozessentwicklungslauf | Frühe Herstellung | Generierung erster Modellgewichte durch iterative Trainingsläufe (iterativ mit Evaluierungen in vordefinierten Intervallen) |
| Formulierung & Fill Finish zur Erstellung des ersten Arzneimittels | Training (Testläufe) | Benutzeroberfläche mit Modell verbunden |
| Präklinisch (mehrere In-vitro, Tiere), Phase-0-Studien. Ethische Genehmigung | Frühe Arzneimittelbewertung und iterative Verbesserung in kontrollierten Umgebungen | |
| Toxikologie. Beginn der Bewertung von Eigenschaften, Sicherheit und Wirksamkeit in hochkontrollierter, risikoarmer Umgebung | Sicherheitsbewertungen | Sicherheitsbewertungen gefährlicher Fähigkeiten (Kontrollierbarkeit, autonome Replikation, Täuschung usw.) via Standardtests und Red-Teaming |
| Pharmakologie (Pharmakodynamik & Kinetik) | Interpretierbarkeitsläufe, inkl. Fähigkeitstests | |
| Mehrere Modellorganismen | Fairness-Bewertung | |
| Verschiedene Temperaturen, Zeitpunkte und Analysemethoden | Stabilitätstests | Genauigkeits-, Robustheits- und Jailbreaking-Tests |
| Auswertung generierter Daten durch Fachexperten. Proof of Concept etabliert - zusätzliche Investitionen in Skalierung | Beratung mit externen Experten | Evaluierungen durch externe Experten |
| Klinische Charge | Produktions-Herstellungslauf | Modell mit zusätzlichen Daten aus Evaluierungsläufen trainiert, das nun alle Anforderungen erfüllt. Model Fine Tuning |
| Regulatorische und ethische Genehmigung (für jede Studie einzeln) | Umfassende Real-World-Arzneimittelbewertung: Klinische Studien in kontrollierten Umgebungen | Keine kontrollierte Real-World-Evaluierung als Teil des KI-Modellentwicklungsprozesses |
| Phase 1 (~2x): Bewertung der Sicherheit und Dosierung des Medikaments in einer kleinen Gruppe gesunder Freiwilliger. Dies hilft, den sicheren Dosierungsbereich zu bestimmen und potenzielle Nebenwirkungen zu identifizieren. | Beginn der Charakterisierungs-, Sicherheits- und Wirksamkeitsprüfung in einer hochkontrollierten Hochrisiko-Umgebung | |
| Phase 2 (~2x): Bewertung der Wirksamkeit des Medikaments und weitere Beurteilung der Sicherheit in einer größeren Gruppe von Patienten mit der zu behandelnden Erkrankung. | ||
| Phase 3 (~3x): Prüfung des Medikaments in einer noch größeren Patientengruppe zur Bestätigung der Wirksamkeit, Vergleich mit gängigen Behandlungen und Überwachung von Nebenwirkungen in einer vielfältigeren Population. | ||
| Audits während des gesamten Entwicklungsprozesses proportional zur Kritikalität der bereitgestellten Materialien oder Dienstleistungen. Alle notwendigen Sicherheits- und Wirksamkeitsdaten gesammelt und bereit zur Bewertung durch Regulierungsbehörden | Audits und Inspektionen durch Sponsor und Regulierungsbehörden | |
| Antrag | Zulassung & Post-Market-Arzneimittelüberwachung | Schrittweise öffentliche Einführung |
| Phase 4 (~3x): Zugang für die Öffentlichkeit | Beta-Testprogramme | |
| Vollständige Marktfreigabe | Öffentliche Veröffentlichung | |
| Meldung unerwünschter Ereignisse | Rücknahme | |
| Zugangskontrolle | ||
| Neuer Antragsprozess mit neu erhobenen Daten | Aktualisierte Versionen | Bereitstellung eines aktualisierten Modells. Updates |
Übersicht der Sicherheitsprinzipien in klinischen Studien
| Kategorie | Beschreibung | Dokumente |
|---|---|---|
| Vorbereitung | Forschung vor Real-World-Tests: Literatur-, Tier- und später Humandaten, die auf besorgniserregende Effekte und deren Häufigkeit hinweisen, werden in kontinuierlich aktualisierten Berichten konsolidiert, die plausibel verwandte unerwünschte Ereignisse zur Überwachung definieren. | Internationale Richtlinien für verschiedene präklinische Sicherheitstests. Beispiel (obligatorische) präklinische Sicherheitsbroschüre und Beipackzettel (Verbraucher) (Arzt) Pfizer / BioNTech Comirnaty. |
| Optimierte experimentelle Designs: Einsatz von Strategien wie sequentiellen Tests, Simulationen und Fallzahlberechnungen zur Minimierung von Schäden, Gewährleistung von Ressourceneffizienz und Erkennung unerwünschter Ereignisse bei bestimmten Häufigkeiten (z.B. 1 unerwünschtes Ereignis pro 10.000.000 Benutzerinteraktionen). | FDA und EMA Leitlinien zu statistischen Prinzipien für klinische Studien basierend auf ICH E9 Richtlinie. | |
| Optimierte Erkennungsumgebungen: Spezialisierte Zentren mit Personal, das eine obligatorische Schulung für optimale Erkennung, Reaktion und Meldung unerwünschter Ereignisse hat. | Siehe Abschnitt 4 der Good Clinical Practice Richtlinien und FDA-Leitlinien zu Arztverantwortlichkeiten. | |
| Erkennung | Dedizierte Experimente: Klinische Studien, insbesondere Phase-1-, Phase-3- und Phase-4-Studien, sind groß angelegte Experimente, die gezielt zur Erkennung unerwünschter Ereignisse konzipiert sind. | Internationale Richtlinien für die Sicherheit bei Humanstudien (siehe Abschnitt E1 und E2A-F) und FDA-Leitlinien für Post-Market-Evaluierung. |
| Kontinuierliche Bewertungen unerwünschter Ereignisse: Umfassende strukturierte Datenerhebung während der Entwicklung und nach Marktzugang. Automatisierte Signalerkennung wird häufig durch Pharmakovigilanz implementiert. | ”Pharmakovigilanz ist die Wissenschaft und die Aktivitäten zur Erkennung, Bewertung, zum Verständnis und zur Prävention unerwünschter Wirkungen oder anderer arzneimittel-/impfstoffbezogener Probleme.” WHO-Definition | |
| Aufzeichnung aller negativen Ereignisse: Sicherstellung einer ganzheitlichen Sicht durch Aufzeichnung aller unerwarteten, unerwünschten oder schädlichen Vorkommnisse, unabhängig von ihrer kausalen Beziehung. | ”Ein unerwünschtes Ereignis (AE) kann daher jedes ungünstige und unbeabsichtigte Zeichen sein (einschließlich z.B. eines abnormen Laborbefundes), Symptom oder Krankheit, die zeitlich mit der Anwendung eines Arzneimittels verbunden ist, ob als arzneimittelbezogen betrachtet oder nicht.” ICH E2A | |
| Pflege zuverlässiger Daten: Einsatz prüfbarer Sicherheitsdatenbanken, Einhaltung guter Dokumentationspraktiken und Implementierung verschiedener Formen der Überwachung. | Good Documentation Practice (GDocP) wie in Abschnitt 4.9 der GCP beschrieben. | |
| Management | Kategorisierung und Priorisierung unerwünschter Ereignisse: Zusammenhang (kausale Beziehung zwischen Behandlung und Wirkung), Schwere (Intensität der Erfahrung der betroffenen Person), Erwartbarkeit (Übereinstimmung mit erwarteten unerwünschten Ereignissen), Ergebnis (Schwere der Konsequenzen), Cluster (hierarchische Zuordnung von Ereignissen zu Clustern). | Internationale Richtlinien zum klinischen Sicherheitsdatenmanagement: Definitionen und Standards für die Berichterstattung und die FDA-Umsetzung. MedDRA bietet eine Zuordnung von über 70.000 medizinischen Begriffen. |
| Vordefinierte Ereignisbehandlung: Typspezifische vordefinierte Verfahren und Benachrichtigungen. | Sicherheitsereignisverfahren für Ärzte und Pharmaunternehmen (GCP Abschnitt 4.11, 5.16, 5.17). | |
| Unvoreingenommene Bewertung: Einsatz unabhängiger Monitore/Sicherheitsausschüsse und Mehrparteien-Entscheidungsfindung zur Eliminierung von Interessenkonflikten. | FDA zu Einrichtung und Betrieb von Data Monitoring Committees. |
Dokumente in einem typischen klinischen Studienantrag
-
Der Mindestumfang an Dokumenten umfasst
-
Studienprotokoll
Einleitung (inkl. Studienrationale, Hintergrund, Risiko/Nutzen-Bewertung)
-
Ziele und Endpunkte
-
Studiendesign (inkl. wissenschaftliche Rationale, Ende-der-Studie-Definition, Abbruchkriterien)
-
Studienobjekt (inkl. Ein-/Ausschlusskriterien)
-
KI-Modell-Interventionen und -Management (inkl. Dateninfrastruktur, Verantwortlichkeit, Vorbereitung usw.)
-
Abbruch der Studie
-
Studienbewertungen und -verfahren (inkl. Benchmarks, Bewertungen, Interpretierbarkeitsarbeit, Definition unerwünschter Ereignisse, unabhängiges Monitoring usw.)
-
Statistische Überlegungen (inkl. statistische Hypothesen, Fallzahlbestimmung, Endpunktanalyse usw.)
-
Administrative Angelegenheiten (inkl. Ethik, Datenschutz, informierte Einwilligung, Aufzeichnungen usw.)
-
Prüferbroschüre (ähnlich wie Model Cards in der KI)
-
Informierte Einwilligung
-
Evaluierungsplan für Anbieter
-
Monitoringplan
-
Projektmanagementplan (inkl. Kommunikation, Eskalation)
-
Anbietermanagementplan
-
Trial Master File Plan (umfasst Investigator Site Files)
-
Sicherheitsmanagementplan
-
Risikobewertung und -kategorisierung
-
Datenmanagementplan
-
Statistischer Analyseplan